Tubulin Resists Degradation by Cereblon-Recruiting PROTACs

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Drug Treatments
2.2. Western Blotting and Tubulin Partitioning
2.3. General Chemistry Methods
2.4. Docking and Linker Length Analysis
3. Results
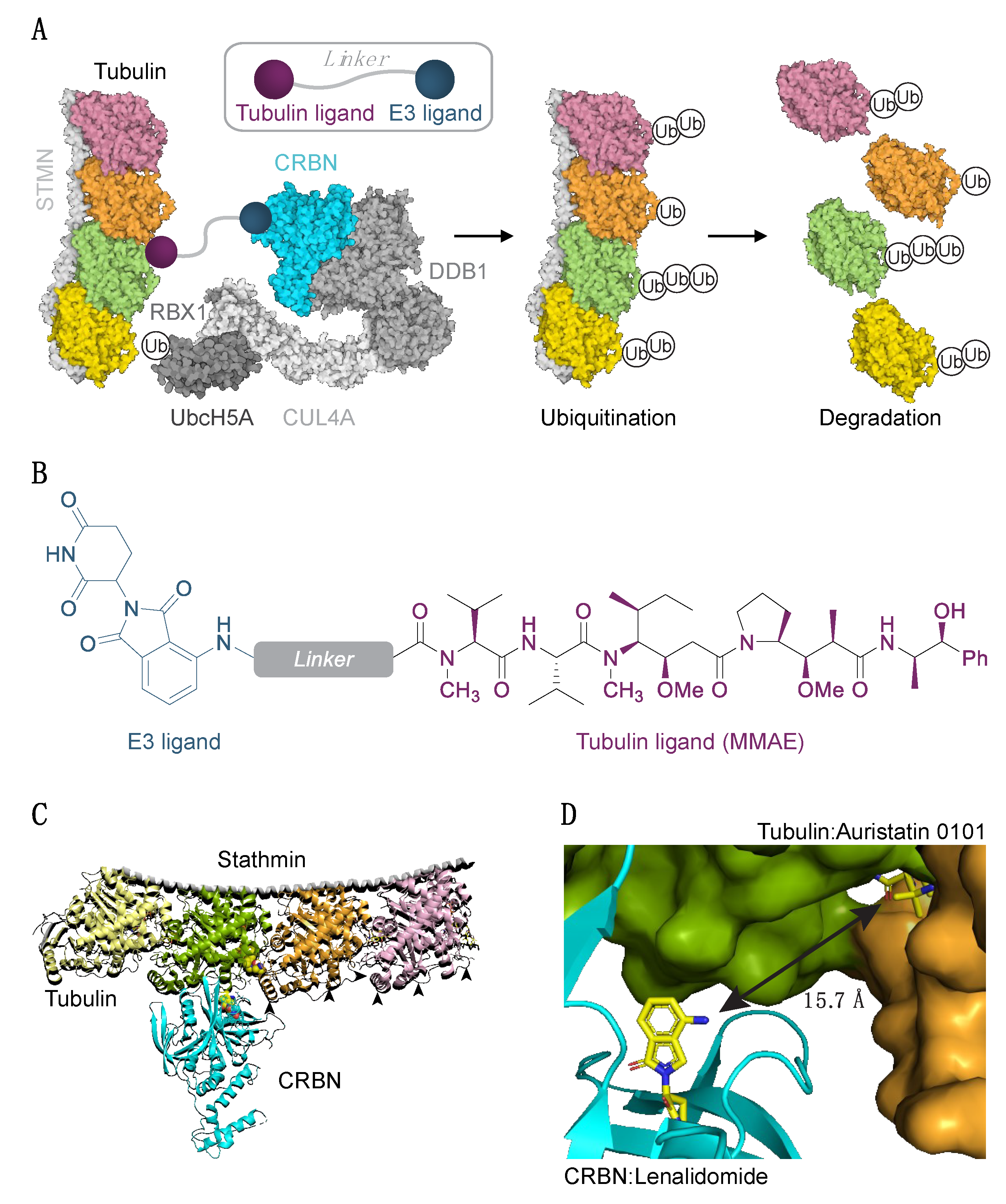
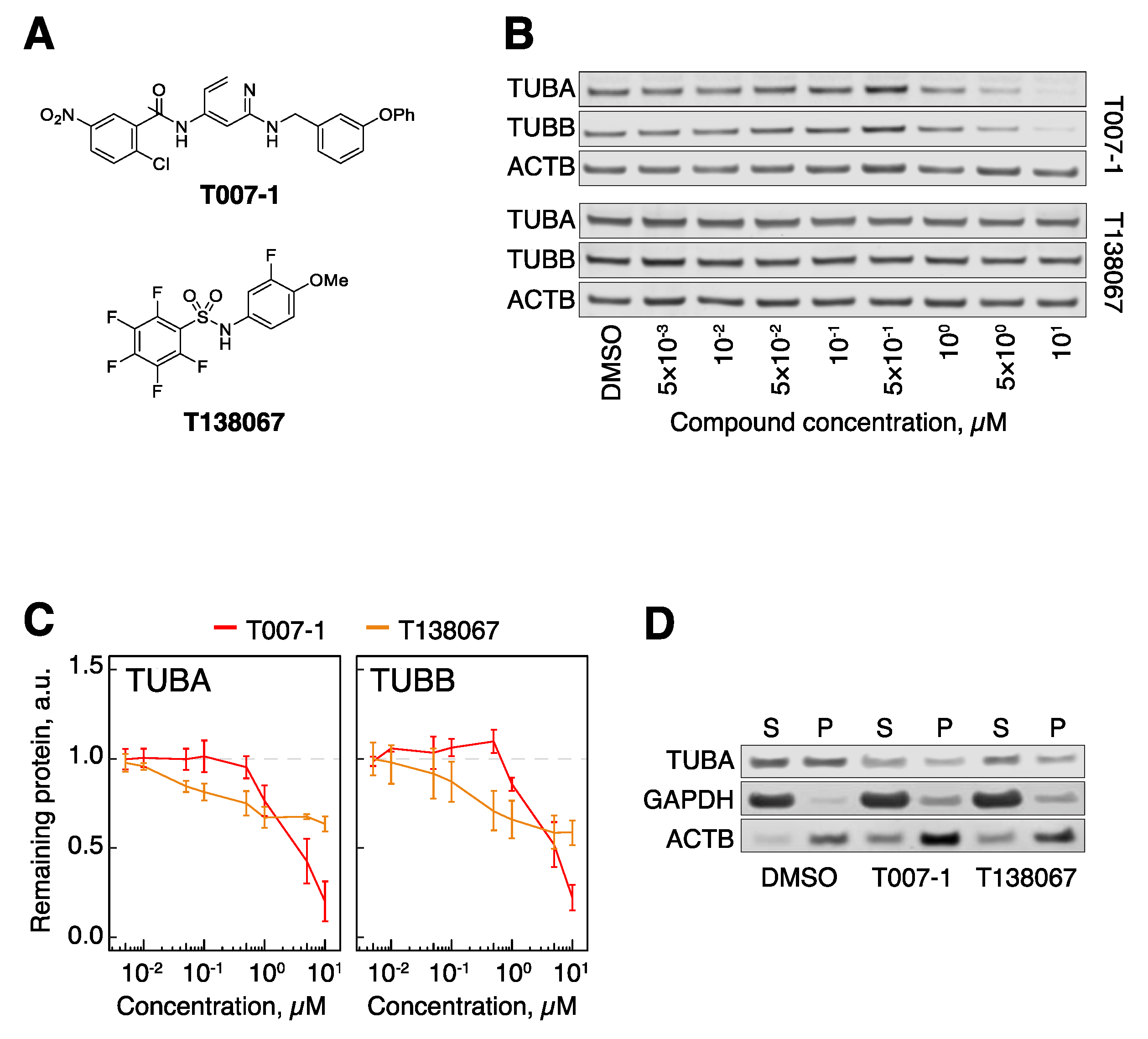
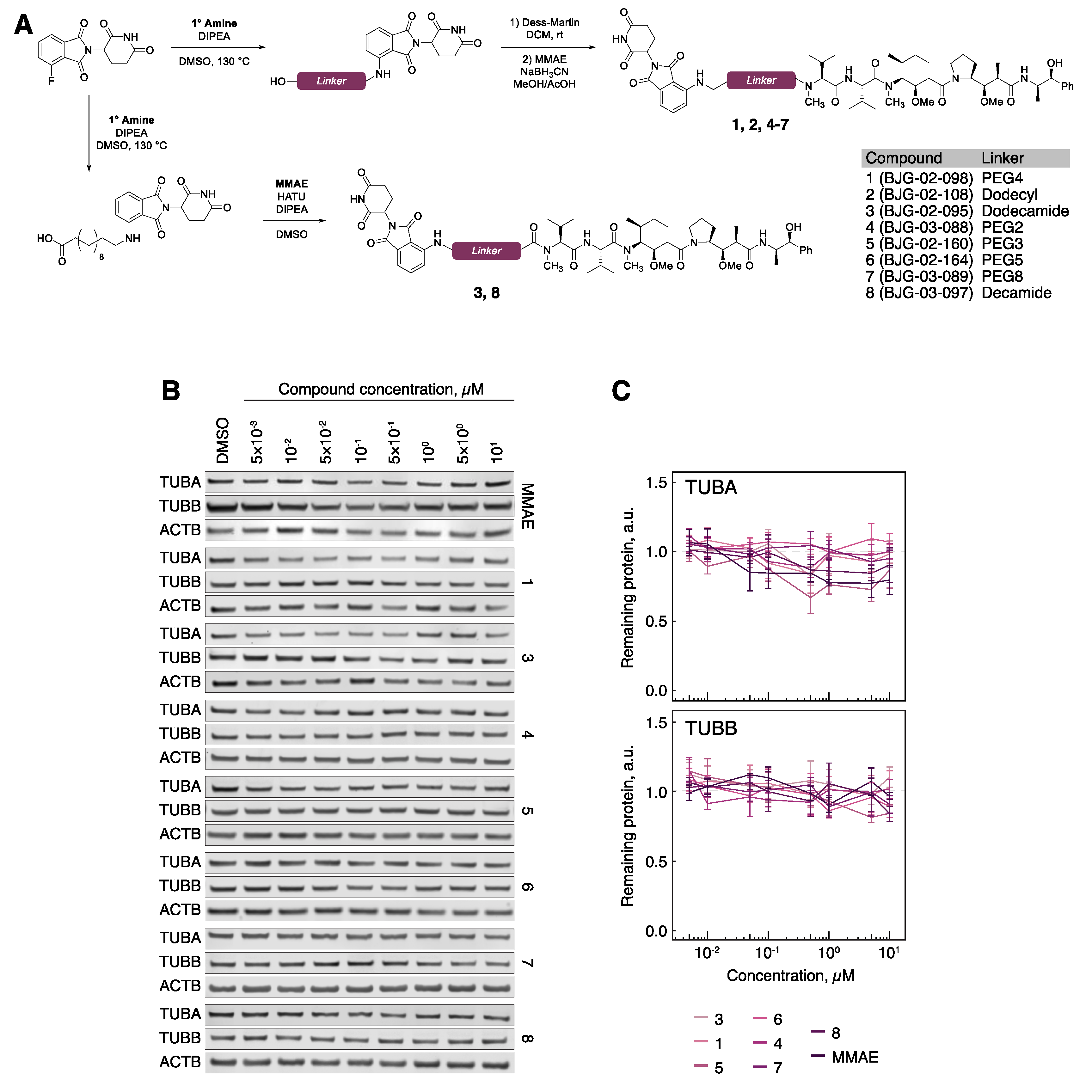
3.1. Cereblon-Based PROTACs with Auristatin Scaffold
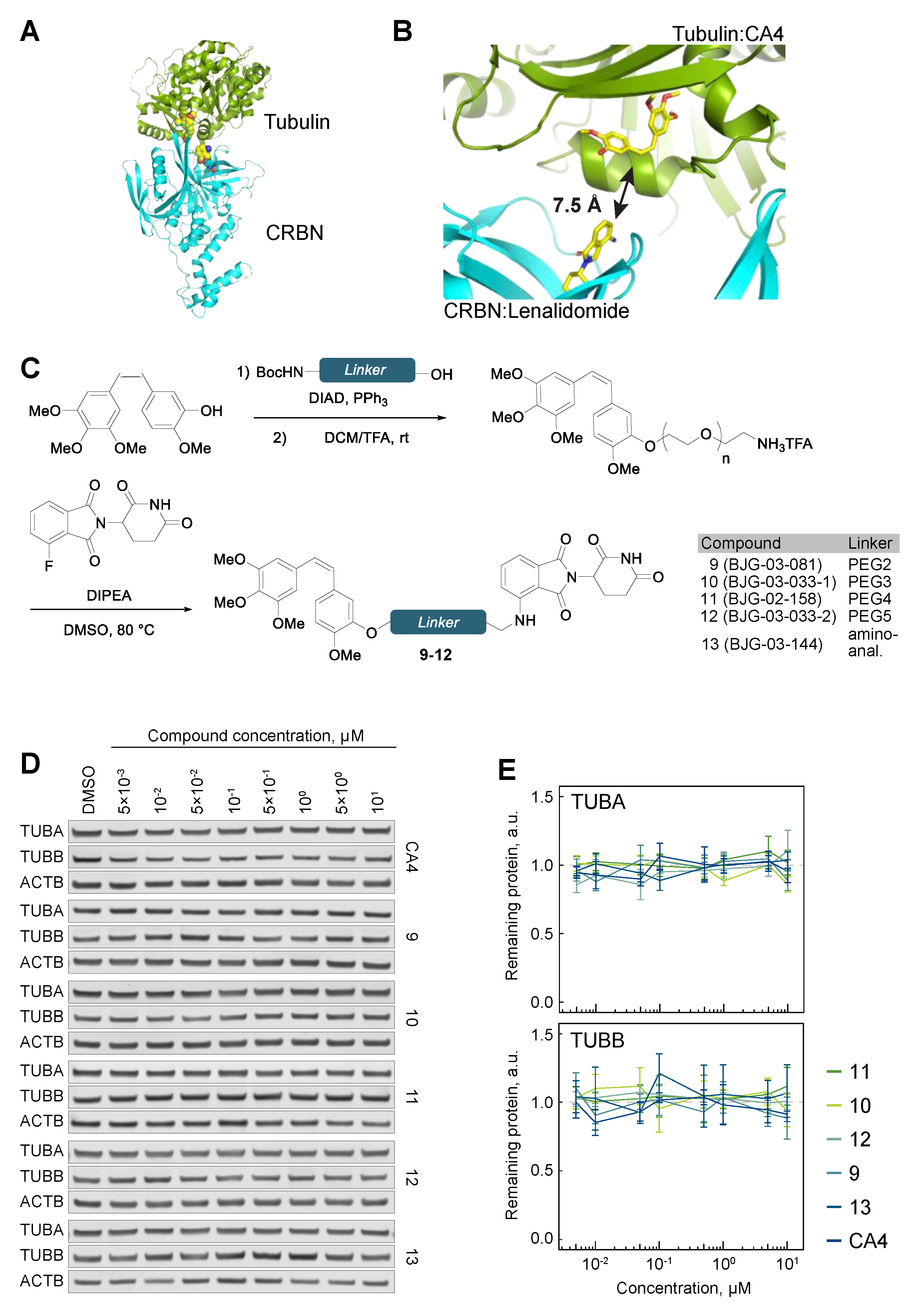
3.2. Cereblon-Based PROTACs with the Combretastatin A-4 Scaffold
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| a.u. | arbitrary units |
| CA4 | combretastatin A-4 |
| CRBN | cereblon |
| CUL4A | cullin 4A |
| DCC | dicyclohexyl carbodiimide |
| DDB1 | damage specific DNA binding protein |
| DMP | Dess-Martin periodinane |
| DMSO | dimethyl sulfoxide |
| HATU | hexafluorophosphate azabenzotriazole tetramethyl uronium |
| hTert | human telomerase reverse transcriptase |
| IC | inhibitory concentration |
| KRAS | Ki-ras2 Kirsten rat sarcoma viral oncogene homolog |
| MMAE | monomethyl auristatin E |
| MOA | mechanism of action |
| P | polymerized |
| PDB | protein data bank |
| PROTAC | proteolysis-targeting chimera |
| RBX1 | ring box 1 |
| RPE1 | retinal pigment epithelium 1 |
| S | soluble |
| SAR | structure-activity relationship |
| SEM | standard error of the mean |
| STMN | stathmin |
| TFA | trifluoroacetic acid |
| TLC | thin-layer chromatography |
| TUBA | α-tubulin |
| TUBB | β-tubulin |
| UbcH5A | ubiquitin-conjugated enzyme E2D1 |
| Ub | ubiquitin |
| UPS | ubiquitin-proteasome system |
References
- Desai, A.; Mitchison, T.J. Microtubule polymerization dynamics. Annu. Rev. Cell Dev. Biol. 1997, 13, 83–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gasic, I.; Mitchison, T.J. Autoregulation and repair in microtubule homeostasis. Curr. Opin. Cell Biol. 2019, 56, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Gasic, I.; Boswell, S.A.; Mitchison, T.J. Tubulin mRNA stability is sensitive to change in microtubule dynamics caused by multiple physiological and toxic cues. PLoS Biol. 2019, 17, e3000225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Z.; Gasic, I.; Chandrasekaran, V.; Peters, N.; Shao, S.; Mitchison, T.J.; Hegde, R.S. TTC5 mediates autoregulation of tubulin via mRNA degradation. Science 2020, 367, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Muroyama, A.; Lechler, T. Microtubule organization, dynamics and functions in differentiated cells. Development 2017, 144, 3012–3021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walczak, C.E.; Heald, R. Mechanisms of mitotic spindle assembly and function. Int. Rev. Cytol. 2008, 265, 111–158. [Google Scholar] [PubMed]
- Ilan, Y. Microtubules: From understanding their dynamics to using them as potential therapeutic targets. J. Cell. Physiol. 2019, 234, 7923–7937. [Google Scholar] [CrossRef] [Green Version]
- Kingston, D.G.I. Tubulin-interactive natural products as anticancer agents. J. Nat. Prod. 2009, 72, 507–515. [Google Scholar] [CrossRef] [Green Version]
- Florian, S.; Mitchison, T.J. Anti-Microtubule Drugs. Methods Mol. Biol. 2016, 1413, 403–421. [Google Scholar]
- Cormier, A.; Marchand, M.; Ravelli, R.B.G.; Knossow, M.; Gigant, B. Structural insight into the inhibition of tubulin by vinca domain peptide ligands. EMBO Rep. 2008, 9, 1101–1106. [Google Scholar] [CrossRef] [Green Version]
- Bai, R.L.; Pettit, G.R.; Hamel, E. Binding of dolastatin 10 to tubulin at a distinct site for peptide antimitotic agents near the exchangeable nucleotide and vinca alkaloid sites. J. Biol. Chem. 1990, 265, 17141–17149. [Google Scholar] [PubMed]
- Cormier, A.; Knossow, M.; Wang, C.; Gigant, B. The binding of vinca domain agents to tubulin: Structural and biochemical studies. Methods Cell Biol. 2010, 95, 373–390. [Google Scholar] [PubMed]
- Hastie, S.B. Interactions of colchicine with tubulin. Pharmacol. Ther. 1991, 51, 377–401. [Google Scholar] [CrossRef]
- Lin, C.M.; Singh, S.B.; Chu, P.S.; Dempcy, R.O.; Schmidt, J.M.; Pettit, G.R.; Hamel, E. Interactions of tubulin with potent natural and synthetic analogs of the antimitotic agent combretastatin: A structure-activity study. Mol. Pharmacol. 1988, 34, 200–208. [Google Scholar] [PubMed]
- Rao, S.; He, L.; Chakravarty, S.; Ojima, I.; Orr, G.A.; Horwitz, S.B. Characterization of the Taxol binding site on the microtubule. Identification of Arg(282) in beta-tubulin as the site of photoincorporation of a 7-benzophenone analogue of Taxol. J. Biol. Chem. 1999, 274, 37990–37994. [Google Scholar] [CrossRef] [Green Version]
- Schiff, P.B.; Fant, J.; Horwitz, S.B. Promotion of microtubule assembly in vitro by taxol. Nature 1979, 277, 665–667. [Google Scholar] [CrossRef]
- Jordan, M.A.; Wilson, L. Microtubules as a target for anticancer drugs. Nat. Rev. Cancer 2004, 4, 253–265. [Google Scholar] [CrossRef]
- Fojo, A.T.; Menefee, M. Microtubule targeting agents: Basic mechanisms of multidrug resistance (MDR). Semin. Oncol. 2005, 32, S3–S8. [Google Scholar] [CrossRef]
- Matamoros, A.J.; Baas, P.W. Microtubules in health and degenerative disease of the nervous system. Brain Res. Bull. 2016, 126, 217–225. [Google Scholar] [CrossRef] [Green Version]
- Cromm, P.M.; Crews, C.M. Targeted Protein Degradation: From Chemical Biology to Drug Discovery. Brain Res. Bull. 2017, 24, 1181–1190. [Google Scholar] [CrossRef] [Green Version]
- Neklesa, T.; Snyder, L.B.; Willard, R.R.; Vitale, N.; Pizzano, J.; Gordon, D.A.; Bookbinder, M.; Macaluso, J.; Dong, H.; Ferraro, C.; et al. ARV-110: An oral androgen receptor PROTAC degrader for prostate cancer. JCO 2019, 37, 259. [Google Scholar] [CrossRef] [Green Version]
- Flanagan, J.J.; Qian, Y.; Gough, S.M.; Andreoli, M.; Bookbinder, M.; Cadelina, G.; Bradley, J.; Rousseau, E.; Willard, R.; Pizzano, J.; et al. Abstract P5-04-18: ARV-471, an oral estrogen receptor PROTAC degrader for breast cancer. Cancer Res. 2019, 79, P5-04-18. [Google Scholar]
- Winter, G.E.; Buckley, D.L.; Paulk, J.; Roberts, J.M.; Souza, A.; Dhe-Paganon, S.; Bradner, J.E. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 2015, 348, 1376–1381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nowak, R.P.; DeAngelo, S.L.; Buckley, D.; He, Z.; Donovan, K.A.; An, J.; Safaee, N.; Jedrychowski, M.P.; Ponthier, C.M.; Ishoey, M.; et al. Plasticity in binding confers selectivity in ligand-induced protein degradation. Nat. Chem. Biol. 2018, 14, 706–714. [Google Scholar] [CrossRef]
- Ohoka, N.; Okuhira, K.; Ito, M.; Nagai, K.; Shibata, N.; Hattori, T.; Ujikawa, O.; Shimokawa, K.; Sano, O.; Koyama, R.; et al. In Vivo Knockdown of Pathogenic Proteins via Specific and Nongenetic Inhibitor of Apoptosis Protein (IAP)-dependent Protein Erasers (SNIPERs). J. Biol. Chem. 2017, 292, 4556–4570. [Google Scholar] [CrossRef] [Green Version]
- Shibata, N.; Nagai, K.; Morita, Y.; Ujikawa, O.; Ohoka, N.; Hattori, T.; Koyama, R.; Sano, O.; Imaeda, Y.; Nara, H.; et al. Development of Protein Degradation Inducers of Androgen Receptor by Conjugation of Androgen Receptor Ligands and Inhibitor of Apoptosis Protein Ligands. J. Med. Chem. 2018, 61, 543–575. [Google Scholar] [CrossRef]
- De Wispelaere, M.; Du, G.; Donovan, K.A.; Zhang, T.; Eleuteri, N.A.; Yuan, J.C.; Kalabathula, J.; Nowak, R.P.; Fischer, E.S.; Gray, N.S.; et al. Small molecule degraders of the hepatitis C virus protease reduce susceptibility to resistance mutations. Nat. Commun. 2019, 10, 3468. [Google Scholar] [CrossRef] [Green Version]
- Yang, K.; Song, Y.; Xie, H.; Wu, H.; Wu, Y.-T.; Leisten, E.D.; Tang, W. Development of the first small molecule histone deacetylase 6 (HDAC6) degraders. Bioorganic Med. Chem. Lett. 2018, 28, 2493–2497. [Google Scholar] [CrossRef]
- Silva, M.C.; Ferguson, F.M.; Cai, Q.; Donovan, K.A.; Nandi, G.; Patnaik, D.; Zhang, T.; Huang, H.-T.; Lucente, D.E.; Dickerson, B.C.; et al. Targeted degradation of aberrant tau in frontotemporal dementia patient-derived neuronal cell models. Elife 2019, 8, 789. [Google Scholar] [CrossRef]
- Lai, A.C.; Toure, M.; Hellerschmied, D.; Salami, J.; Jaime-Figueroa, S.; Ko, E.; Hines, J.; Crews, C.M. Modular PROTAC Design for the Degradation of Oncogenic BCR-ABL. Angew. Chem. Int. Ed. 2016, 55, 807–810. [Google Scholar] [CrossRef] [Green Version]
- Powell, C.E.; Gao, Y.; Tan, L.; Donovan, K.A.; Nowak, R.P.; Loehr, A.; Bahcall, M.; Fischer, E.S.; Jänne, P.A.; George, R.E.; et al. Chemically Induced Degradation of Anaplastic Lymphoma Kinase (ALK). J. Med. Chem. 2018, 61, 4249–4255. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.; Wang, E.S.; Donovan, K.A.; Liang, Y.; Fischer, E.S.; Zhang, T.; Gray, N.S. Development of Dual and Selective Degraders of Cyclin-Dependent Kinases 4 and 6. Angew. Chem. Int. Ed. 2019, 58, 6321–6326. [Google Scholar] [CrossRef] [PubMed]
- Popow, J.; Arnhof, H.; Bader, G.; Berger, H.; Ciulli, A.; Covini, D.; Dank, C.; Gmaschitz, T.; Greb, P.; Karolyi-Özguer, J.; et al. Highly Selective PTK2 Proteolysis Targeting Chimeras to Probe Focal Adhesion Kinase Scaffolding Functions. J. Med. Chem. 2019, 62, 2508–2520. [Google Scholar] [CrossRef] [PubMed]
- Mi, L.; Gan, N.; Cheema, A.; Dakshanamurthy, S.; Wang, X.; Yang, D.C.H.; Chung, F.-L. Cancer preventive isothiocyanates induce selective degradation of cellular alpha- and beta-tubulins by proteasomes. J. Biol. Chem. 2009, 284, 17039–17051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Li, Y.; Yan, W.; Li, W.; Qiu, Q.; Ye, H.; Chen, L. Covalent modification of Cys-239 in β-tubulin by small molecules as a strategy to promote tubulin heterodimer degradation. J. Biol. Chem. 2019, 294, 8161–8170. [Google Scholar] [CrossRef] [PubMed]
- Harris, G.; Schaefer, K.L. The microtubule-targeting agent T0070907 induces proteasomal degradation of tubulin. Biochem. Biophys. Res. Commun. 2009, 388, 345–349. [Google Scholar] [CrossRef] [PubMed]
- Goodsell, D.S.; Autin, L.; Olson, A.J. Illustrate: Software for Biomolecular Illustration. Structure 2019, 27, 1716–1720.e1. [Google Scholar] [CrossRef]
- Morin, A.; Eisenbraun, B.; Key, J.; Sanschagrin, P.C.; Timony, M.A.; Ottaviano, M.; Sliz, P. Collaboration gets the most out of software. Elife 2013, 2, e01456. [Google Scholar] [CrossRef]
- Li, Z.; Pinch, B.J.; Olson, C.M.; Donovan, K.A.; Nowak, R.P.; Mills, C.E.; Scott, D.A.; Doctor, Z.M.; Eleuteri, N.A.; Chung, M.; et al. Development and Characterization of a Wee1 Kinase Degrader. Cell Chem. Biol. 2020, 27, 57–65.e9. [Google Scholar] [CrossRef]
- Varetti, G.; Musacchio, A. The spindle assembly checkpoint. Curr. Biol. 2008, 18, R591–R595. [Google Scholar] [CrossRef] [Green Version]
- Cirla, A.; Mann, J. Combretastatins: From natural products to drug discovery. Nat. Prod. Rep. 2003, 20, 558–564. [Google Scholar] [CrossRef] [PubMed]
- Pettit, G.R.; Singh, S.B.; Niven, M.L.; Hamel, E.; Schmidt, J.M. Isolation, structure, and synthesis of combretastatins A-1 and B-1, potent new inhibitors of microtubule assembly, derived from Combretum caffrum. ACS Publ. 1987, 50, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, V.; Venghateri, J.B.; Dhaked, H.P.S.; Bhoyar, A.S.; Guchhait, S.K.; Panda, D. Novel Combretastatin-2-aminoimidazole Analogues as Potent Tubulin Assembly Inhibitors: Exploration of Unique Pharmacophoric Impact of Bridging Skeleton and Aryl Moiety. J. Med. Chem. 2016, 59, 3439–3451. [Google Scholar] [CrossRef] [PubMed]
- Pettit, G.R.; Rhodes, M.R.; Herald, D.L.; Hamel, E.; Schmidt, J.M.; Pettit, R.K. Antineoplastic Agents. 445. Synthesis and Evaluation of Structural Modifications of (Z)- and (E)-Combretastatin A-4 | Journal of Medicinal Chemistry. J. Med. Chem. 2005, 48, 4087–4099. [Google Scholar] [CrossRef]
- Lewis, S.A.; Tian, G.; Cowan, N.J. The α-and β-tubulin folding pathways. Trends Cell Biol. 1997, 7, 479–484. [Google Scholar] [CrossRef]
- Mitchison, T.; Kirschner, M. Dynamic instability of microtubule growth. Nature 1984, 312, 237–242. [Google Scholar] [CrossRef]
- Caron, J.M.; Jones, A.L.; Kirschner, M.W. Autoregulation of tubulin synthesis in hepatocytes and fibroblasts. J. Cell Biol. 1985, 101, 1763–1772. [Google Scholar] [CrossRef]
- Cleveland, D.W.; Lopata, M.A.; Sherline, P.; Kirschner, M.W. Unpolymerized tubulin modulates the level of tubulin mRNAs. Cell 1981, 25, 537–546. [Google Scholar] [CrossRef]
- Zeng, M.; Xiong, Y.; Safaee, N.; Nowak, R.P.; Donovan, K.A.; Yuan, C.J.; Nabet, B.; Gero, T.W.; Feru, F.; Li, L.; et al. Exploring Targeted Degradation Strategy for Oncogenic KRASG12C. Cell Chem. Biol. 2020, 27, 19–31.e6. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gasic, I.; Groendyke, B.J.; Nowak, R.P.; Yuan, J.C.; Kalabathula, J.; Fischer, E.S.; Gray, N.S.; Mitchison, T.J. Tubulin Resists Degradation by Cereblon-Recruiting PROTACs. Cells 2020, 9, 1083. https://doi.org/10.3390/cells9051083
Gasic I, Groendyke BJ, Nowak RP, Yuan JC, Kalabathula J, Fischer ES, Gray NS, Mitchison TJ. Tubulin Resists Degradation by Cereblon-Recruiting PROTACs. Cells. 2020; 9(5):1083. https://doi.org/10.3390/cells9051083
Chicago/Turabian StyleGasic, Ivana, Brian J. Groendyke, Radosław P. Nowak, J. Christine Yuan, Joann Kalabathula, Eric S. Fischer, Nathanael S. Gray, and Timothy J. Mitchison. 2020. "Tubulin Resists Degradation by Cereblon-Recruiting PROTACs" Cells 9, no. 5: 1083. https://doi.org/10.3390/cells9051083
APA StyleGasic, I., Groendyke, B. J., Nowak, R. P., Yuan, J. C., Kalabathula, J., Fischer, E. S., Gray, N. S., & Mitchison, T. J. (2020). Tubulin Resists Degradation by Cereblon-Recruiting PROTACs. Cells, 9(5), 1083. https://doi.org/10.3390/cells9051083