Molecular Mechanisms of Heat Shock Factors in Cancer

Abstract
:1. Introduction
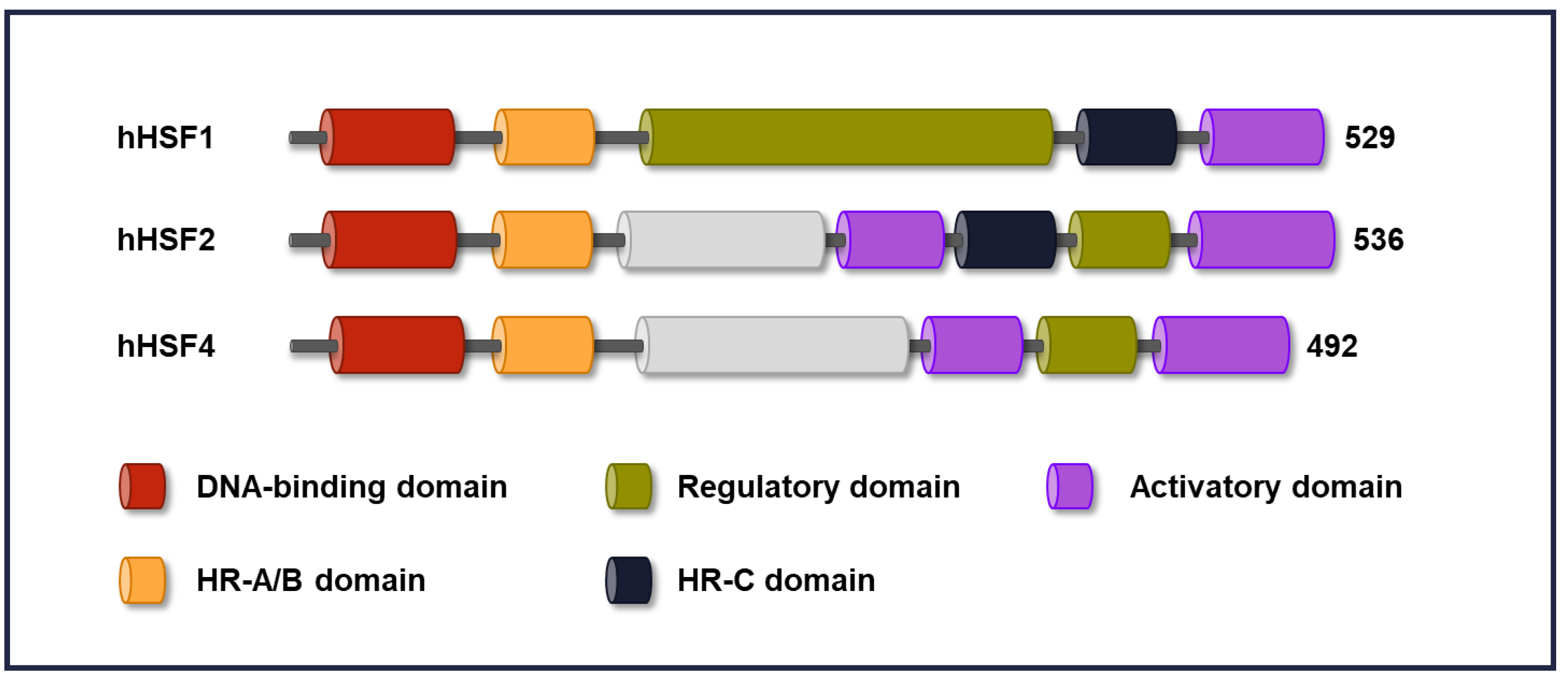
2. Domain Structure in HSFs
3. Activation and Attenuation
3.1. Molecular Mechanisms Regulating HSF Activation
3.2. Molecular Mechanisms Regulating HSF Attenuation
4. HSFs as Developmental Factors
4.1. HSFs in Fertility
4.2. HSFs in Brain Development and Sensory Organs
5. HSFs in Cancer
5.1. HSF1 Cancer Signature
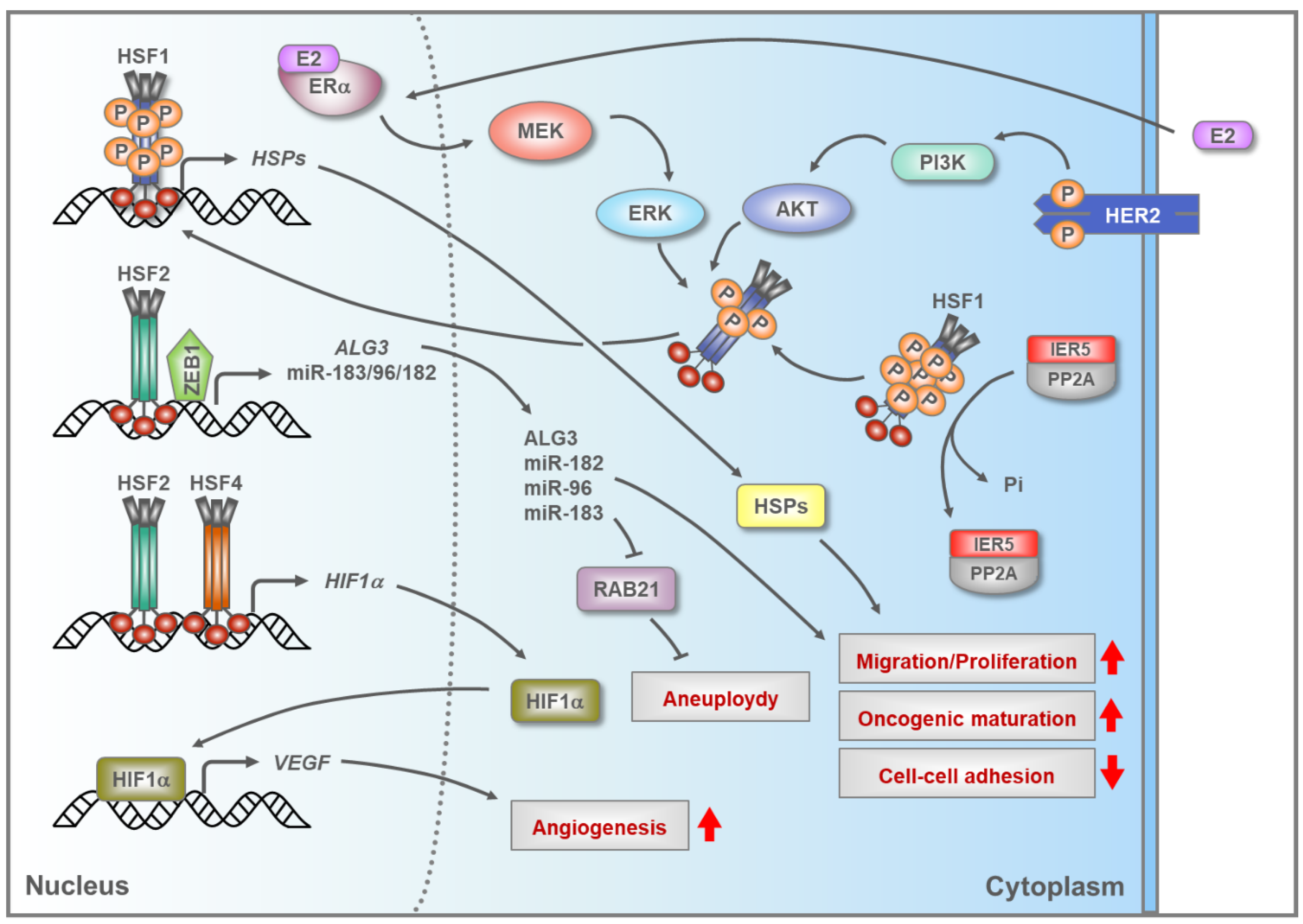
5.2. HSFs in Breast Cancer
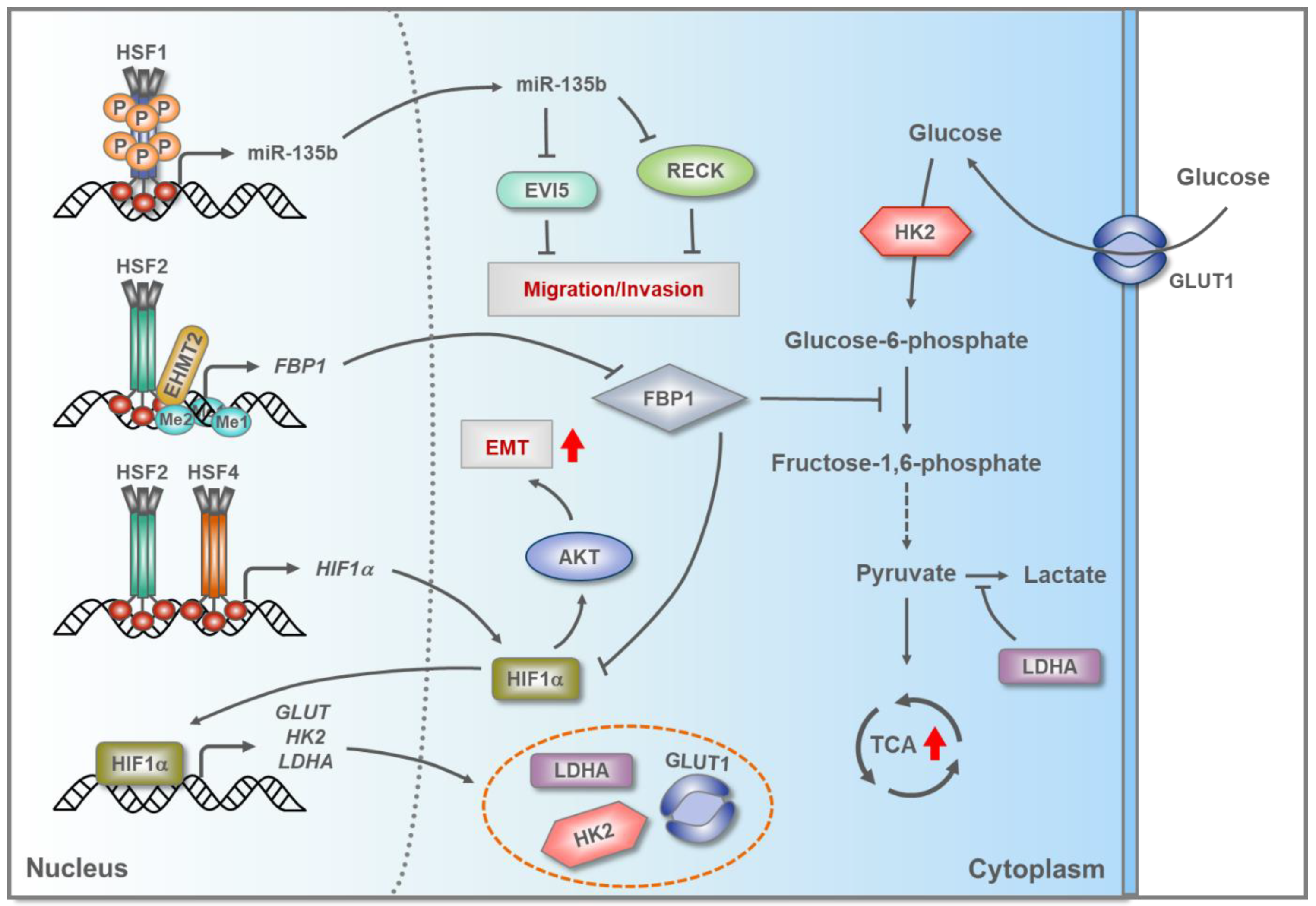
5.3. HSFs in Hepatocellular Carcinoma (HCC)
5.4. HSF1 and HSF2 in Prostate Cancer
5.5. HSF1 and HSF2 in Lung Cancer and Esophageal Squamous Cell Carcinoma (ESCC)
5.6. HSF1 and HSF4 in Colorectal Cancer (CRC)
6. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Abane, R.; Mezger, V. Roles of heat shock factors in gametogenesis and development. FEBS J. 2010, 277, 4150–4172. [Google Scholar] [CrossRef]
- Joutsen, J.; Sistonen, L. Tailoring of proteostasis networks with heat shock factors. Cold Spring Harb. Perspect. Biol. 2019, 11, a034066. [Google Scholar] [CrossRef] [PubMed]
- Ritossa, F. A new puffing pattern induced by temperature shock and DNP in Drosophila. Experientia 1962, 18, 571–573. [Google Scholar] [CrossRef]
- Hartl, F.U.; Bracher, A.; Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 2011, 475, 324–332. [Google Scholar] [CrossRef]
- Schuetz, T.J.; Gallo, G.J.; Sheldon, L.; Tempst, P.; Kingston, R.E. Isolation of a cDNA for HSF2: Evidence for two heat shock factor genes in humans. Proc. Natl. Acad. Sci. USA 1991, 88, 6911–6915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakai, A.; Morimoto, R.I. Characterization of a novel chicken heat shock transcription factor, heat shock factor 3, suggests a new regulatory pathway. Mol. Cell. Biol. 1993, 13, 1983–1997. [Google Scholar] [CrossRef]
- Nakai, A.; Tanabe, M.; Kawazoe, Y.; Inazawa, J.; Morimoto, R.I.; Nagata, K. HSF4, a new member of the human heat shock factor family which lacks properties of a transcriptional activator. Mol. Cell. Biol. 1997, 17, 469–481. [Google Scholar] [CrossRef] [Green Version]
- Vihervaara, A.; Sergelius, C.; Vasara, J.; Blom, M.A.; Elsing, A.N.; Roos-Mattjus, P.; Sistonen, L. Transcriptional response to stress in the dynamic chromatin environment of cycling and mitotic cells. Proc. Natl. Acad. Sci. USA 2013, 110, E3388–E3397. [Google Scholar] [CrossRef] [Green Version]
- Mahat, D.B.; Salamanca, H.H.; Duarte, F.M.; Danko, C.G.; Lis, J.T. Mammalian heat shock response and mechanisms underlying its genome-wide transcriptional regulation. Mol. Cell 2016, 62, 63–78. [Google Scholar] [CrossRef] [Green Version]
- Mendillo, M.L.; Santagata, S.; Koeva, M.; Bell, G.W.; Hu, R.; Tamimi, R.M.; Fraenkel, E.; Ince, T.A.; Whitesell, L.; Lindquist, S. HSF1 drives a transcriptional program distinct from heat shock to support highly malignant human cancers. Cell 2012, 150, 549–562. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Chauve, L.; Phelps, G.; Brielmann, R.M.; Morimoto, R.I. E2F coregulates an essential HSF developmental program that is distinct from the heat-shock response. Genes Dev. 2016, 30, 2062–2075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scharf, K.D.; Berberich, T.; Ebersberger, I.; Nover, L. The plant heat stress transcription factor (Hsf) family: Structure, function and evolution. Biochim. Biophys. Acta 2012, 19, 104–119. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Pastor, R.; Burchfiel, E.T.; Thiele, D.J. Regulation of heat shock transcription factors and their roles in physiology and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 4–19. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Eroglu, B.; Moskophidis, D.; Mivechi, N.F. Targeted deletion of Hsf1, 2, and 4 genes in mice. Methods Mol. Biol. 2018, 1709, 1–22. [Google Scholar] [PubMed] [Green Version]
- Budzyński, M.A.; Sistonen, L. Versatile functions of heat shock factors: It is not all about stress. Curr. Immunol. Rev. 2017, 13. [Google Scholar] [CrossRef]
- Jaeger, A.M.; Makley, L.N.; Gestwicki, J.E.; Thiele, D.J. Genomic heat shock element sequences drive cooperative human heat shock factor 1 DNA binding and selectivity. J. Biol. Chem. 2014, 289, 30459–30469. [Google Scholar] [CrossRef] [Green Version]
- Peteranderl, R.; Rabenstein, M.; Shin, Y.K.; Liu, C.W.; Wemmer, D.E.; King, D.S.; Nelson, H.C. Biochemical and biophysical characterization of the trimerization domain from the heat shock transcription factor. Biochemistry 1999, 38, 3559–3569. [Google Scholar] [CrossRef]
- Östling, P.; Björk, J.K.; Roos-Mattjus, P.; Mezger, V.; Sistonen, L. Heat shock factor 2 (HSF2) contributes to inducible expression of hsp genes through interplay with HSF1. J. Biol. Chem. 2007, 282, 7077–7086. [Google Scholar] [CrossRef] [Green Version]
- Sandqvist, A.; Björk, J.K.; Åkerfelt, M.; Chitikova, Z.; Grichine, A.; Vourc’h, C.; Jolly, C.; Salminen, T.A.; Nymalm, Y.; Sistonen, L. Heterotrimerization of heat-shock factors 1 and 2 provides a transcriptional switch in response to distinct stimuli. Mol. Biol. Cell 2009, 20, 1340–1347. [Google Scholar] [CrossRef] [Green Version]
- Rabindran, S.; Haroun, R.; Clos, J.; Wisniewski, J.; Wu, C. Regulation of heat shock factor trimer formation: Role of a conserved leucine zipper. Science 1993, 259, 230–234. [Google Scholar] [CrossRef]
- Sullivan, E.K.; Weirich, C.S.; Guyon, J.R.; Sif, S.; Kingston, R.E. Transcriptional activation domains of human heat shock factor 1 recruit human SWI/SNF. Mol. Cell. Biol. 2001, 21, 5826–5837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.M.; Werner, J.; Kim, J.M.; Lis, J.T.; Kim, Y.J. Mediator, not holoenzyme, is directly recruited to the heat shock promoter by HSF upon heat shock. Mol. Cell 2001, 8, 9–19. [Google Scholar] [CrossRef]
- Vujanac, M.; Fenaroli, A.; Zimarino, V. Constitutive nuclear import and stress-regulated nucleocytoplasmic shuttling of mammalian heat-shock factor 1. Traffic 2005, 6, 214–229. [Google Scholar] [CrossRef] [PubMed]
- Goodson, M.L.; Sarge, K.D. Heat-inducible DNA binding of purified heat shock transcription factor 1. J. Biol. Chem. 1995, 270, 2447–2450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, S.G.; Thiele, D.J. Redox regulation of mammalian heat shock factor 1 is essential for Hsp gene activation and protection from stress. Genes Dev. 2003, 17, 516–528. [Google Scholar] [CrossRef] [Green Version]
- Hentze, N.; Le Breton, L.; Wiesner, J.; Kempf, G.; Mayer, M.P. Molecular mechanism of thermosensory function of human heat shock transcription factor Hsf1. Elife 2016, 5, e11576. [Google Scholar] [CrossRef]
- Green, M.; Schuetz, T.J.; Sullivan, E.K.; Kingston, R.E. A heat shock responsive domain of human HSF1 that regulates transcription activation domain function. Mol. Cell. Biol. 1995, 15, 3354–3362. [Google Scholar] [CrossRef] [Green Version]
- Knauf, U.; Newton, E.M.; Kyriakis, J.; Kingston, R.E. Repression of human heat shock factor 1 activity at control temperature by phosphorylation. Genes Dev. 1996, 10, 2782–2793. [Google Scholar] [CrossRef] [Green Version]
- Budzyński, M.A.; Puustinen, M.C.; Joutsen, J.; Sistonen, L. Uncoupling stress-inducible phosphorylation of heat shock factor 1 from its activation. Mol. Cell. Biol. 2015, 35, 2530–2540. [Google Scholar] [CrossRef] [Green Version]
- Breton, L.; Mayer, M.P. A model for handling cell stress. Elife 2016, 5, e22850. [Google Scholar] [CrossRef]
- Powers, M.V.; Workman, P. Inhibitors of the heat shock response: Biology and pharmacology. FEBS Lett. 2007, 581, 3758–3769. [Google Scholar] [CrossRef] [Green Version]
- Guisbert, E.; Czyz, D.M.; Richter, K.; McMullen, P.D.; Morimoto, R.I. Identification of a tissue-selective heat shock response regulatory network. PLoS Genet. 2013, 9, e1003466. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.; Krakowiak, J.; Patel, N.; Beyzavi, A.; Ezike, J.; Khalil, A.S.; Pincus, D. Dynamic control of Hsf1 during heat shock by a chaperone switch and phosphorylation. Elife 2016, 5, e18638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krakowiak, J.; Zheng, X.; Patel, N.; Feder, Z.A.; Anandhakumar, J.; Valerius, K.; Gross, D.S.; Khalil, A.S.; Pincus, D. Hsf1 and Hsp70 constitute a two-component feedback loop that regulates the yeast heat shock response. Elife 2018, 7, e31668. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Mosser, D.D.; Morimoto, R.I. Molecular chaperones as HSF1-specific transcriptional repressors. Genes Dev. 1998, 12, 654–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prahlad, V.; Cornelius, T.; Morimoto, R.I. Regulation of the cellular heat shock response in Caenorhabditis elegans by thermosensory neurons. Science 2008, 320, 811–814. [Google Scholar] [CrossRef] [Green Version]
- Tatum, M.C.; Ooi, F.K.; Chikka, M.R.; Chauve, L.; Martinez-Velazquez, L.A.; Steinbusch, H.W.M.; Morimoto, R.I.; Prahlad, V. Neuronal serotonin release triggers the heat shock response in C. elegans in the absence of temperature increase. Curr. Biol. 2015, 25, 163–174. [Google Scholar] [CrossRef] [Green Version]
- Takii, R.; Fujimoto, M.; Tan, K.; Takaki, E.; Hayashida, N.; Nakato, R.; Shirahige, K.; Nakai, A. ATF1 modulates the heat shock response by regulating the stress-inducible heat shock factor 1 transcription complex. Mol. Cell. Biol. 2015, 35, 11–25. [Google Scholar] [CrossRef] [Green Version]
- Fujimoto, M.; Takii, R.; Katiyar, A.; Srivastava, P.; Nakai, A. Poly(ADP-Ribose) polymerase 1 promotes the human heat shock response by facilitating heat shock transcription factor 1 binding to DNA. Mol. Cell. Biol. 2018, 38, e00051-18. [Google Scholar]
- Fujimoto, M.; Takaki, E.; Takii, R.; Tan, K.; Prakasam, R.; Hayashida, N.; Iemura, S.; Natsume, T.; Nakai, A. RPA assists HSF1 access to nucleosomal DNA by recruiting histone chaperone FACT. Mol. Cell 2012, 48, 182–194. [Google Scholar] [CrossRef] [Green Version]
- Sarge, K.D.; Murphy, S.P.; Morimoto, R.I. Activation of heat shock gene transcription by heat shock factor 1 involves oligomerization, acquisition of DNA-binding activity, and nuclear localization and can occur in the absence of stress. Mol. Cell. Biol. 1993, 13, 1392–1407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vihervaara, A.; Duarte, F.M.; Lis, J.T. Molecular mechanisms driving transcriptional stress responses. Nat. Rev. Genet. 2018, 19, 385–397. [Google Scholar] [CrossRef] [PubMed]
- Himanen, S.V.; Sistonen, L. New insights into transcriptional reprogramming during cellular stress. J. Cell Sci. 2019, 132, jcs238402. [Google Scholar] [CrossRef] [PubMed]
- Hantsche, M.; Cramer, P. Conserved RNA polymerase II initiation complex structure. Curr. Opin. Struct. Biol. 2017, 47, 17–22. [Google Scholar] [CrossRef] [Green Version]
- Soutourina, J. Transcription regulation by the mediator complex. Nat. Rev. Mol. Cell Biol. 2018, 19, 262–274. [Google Scholar] [CrossRef]
- Gómez, A.V.; Galleguillos, D.; Maass, J.C.; Battaglioli, E.; Kukuljan, M.; Andrés, M.E. CoREST represses the heat shock response mediated by HSF1. Mol. Cell 2008, 31, 222–231. [Google Scholar] [CrossRef]
- Kijima, T.; Prince, T.L.; Tigue, M.L.; Yim, K.H.; Schwartz, H.; Beebe, K.; Lee, S.; Budzyński, M.A.; Williams, H.; Trepel, J.B.; et al. HSP90 inhibitors disrupt a transient HSP90-HSF1 interaction and identify a noncanonical model of HSP90-mediated HSF1 regulation. Sci. Rep. 2018, 8, 6976. [Google Scholar] [CrossRef] [Green Version]
- Westerheide, S.D.; Anckar, J.; Stevens, S.M., Jr.; Sistonen, L.; Morimoto, R.I. Stress-inducible regulation of heat shock factor 1 by the deacetylase SIRT1. Science 2009, 323, 1063–1066. [Google Scholar] [CrossRef] [Green Version]
- Zelin, E.; Zhang, Y.; Toogun, O.A.; Zhong, S.; Freeman, B.C. The p23 molecular chaperone and GCN5 acetylase jointly modulate protein-DNA dynamics and open chromatin status. Mol. Cell 2012, 48, 459–470. [Google Scholar] [CrossRef] [Green Version]
- Kourtis, N.; Moubarak, R.S.; Aranda-Orgilles, B.; Lui, K.; Aydin, I.T.; Trimarchi, T.; Darvishian, F.; Salvaggio, C.; Zhong, J.; Bhattm, K.; et al. FBXW7 modulates cellular stress response and metastatic potential through HSF1 post-translational modification. Nat. Cell Biol. 2015, 17, 322–332. [Google Scholar] [CrossRef]
- Yang, T.; Ren, C.; Lu, C.; Qiao, P.; Han, X.; Wang, L.; Wang, D.; Lv, S.; Sun, Y.; Yu, Z. Phosphorylation of HSF1 by PIM2 Induces PD-L1 expression and promotes tumor growth in breast cancer. Cancer Res. 2019, 79, 5233–5244. [Google Scholar] [CrossRef] [PubMed]
- Ahlskog, J.K.; Björk, J.K.; Elsing, A.N.; Aspelin, C.; Kallio, M.; Roos-Mattjus, P.; Sistonen, L. Anaphase-promoting complex/cyclosome participates in the acute response to protein damaging stress. Mol. Cell. Biol. 2010, 30, 5608–5620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorger, P.K.; Pelham, H.R. Yeast heat shock factor is an essential DNA-binding protein that exhibits temperature-dependent phosphorylation. Cell 1988, 54, 855–864. [Google Scholar] [CrossRef]
- Jedlicka, P.; Mortin, M.A.; Wu, C. Multiple functions of Drosophila heat shock transcription factor in vivo. EMBO J. 1997, 16, 2452–2462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uchida, S.; Hara, K.; Kobayashi, A.; Fujimoto, M.; Otsuki, K.; Yamagata, H.; Hobara, T.; Abe, N.; Higuchi, F.; Shibata, T.; et al. Impaired hippocampal spinogenesis and neurogenesis and altered affective behavior in mice lacking heat shock factor 1. Proc. Natl. Acad. Sci. USA 2011, 108, 1681–1686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takaki, E.; Fujimoto, M.; Sugahara, K.; Nakahari, T.; Yonemura, S.; Tanaka, Y.; Hayashida, N.; Inouye, S.; Takemoto, T.; Yamashita, H.; et al. Maintenance of olfactory neurogenesis requires HSF1, a major heat shock transcription factor in mice. J. Biol. Chem. 2006, 281, 4931–4937. [Google Scholar] [CrossRef] [Green Version]
- DeBry, R.W.; Seldin, M.F. Human/mouse homology relationships. Genomics 1996, 33, 337–351. [Google Scholar] [CrossRef]
- Kallio, M.; Chang, Y.; Manuel, M.; Alastalo, T.P.; Rallu, M.; Gitton, Y.; Pirkkala, L.; Loones, M.T.; Paslaru, L.; Larney, S.; et al. Brain abnormalities, defective meiotic chromosome synapsis and female subfertility in HSF2 null mice. EMBO J. 2002, 21, 2591–2601. [Google Scholar] [CrossRef] [Green Version]
- Åkerfelt, M.; Henriksson, E.; Laiho, A.; Vihervaara, A.; Rautoma, K.; Kotaja, N.; Sistonen, L. Promoter ChIP-chip analysis in mouse testis reveals Y chromosome occupancy by HSF2. Proc. Natl. Acad. Sci. USA 2008, 105, 11224–11229. [Google Scholar] [CrossRef] [Green Version]
- Björk, J.K.; Sandqvist, A.; Elsing, A.N.; Kotaja, N.; Sistonen, L. miR-18, a member of Oncomir-1, targets heat shock transcription factor 2 in spermatogenesis. Development 2010, 137, 3177–3184. [Google Scholar] [CrossRef] [Green Version]
- Christians, E.; Davis, A.; Thomas, S.; Benjamin, I.J. Maternal effect of Hsf1 on reproductive success. Nature 2000, 407, 693–694. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Zuo, X.; Davis, A.A.; McMillan, D.R.; Curry, B.B.; Richardson, J.A.; Benjamin, I.J. HSF1 is required for extra-embryonic development, postnatal growth and protection during inflammatory responses in mice. EMBO J. 1999, 18, 5943–5952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.; Ying, Z.; Jin, X.; Tu, N.; Zhang, Y.; Phillips, M.; Moskophidis, D.; Mivechi, N.F. Essential requirement for both hsf1 and hsf2 transcriptional activity in spermatogenesis and male fertility. Genesis 2004, 38, 66–80. [Google Scholar] [CrossRef] [PubMed]
- Korfanty, J.; Stokowy, T.; Widlak, P.; Gogler-Piglowska, A.; Handschuh, L.; Podkowiński, J.; Vydra, N.; Naumowicz, A.; Toma-Jonik, A.; Widlak, W. Crosstalk between HSF1 and HSF2 during the heat shock response in mouse testes. Int. J. Biochem. Cell Biol. 2014, 57, 76–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, Y.; Östling, P.; Åkerfelt, M.; Trouillet, D.; Rallu, M.; Gitton, Y.; El Fatimy, R.; Fardeau, V.; Le Crom, S.; Morange, M.; et al. Role of heat-shock factor 2 in cerebral cortex formation and as a regulator of p35 expression. Genes Dev. 2006, 20, 836–847. [Google Scholar] [CrossRef] [Green Version]
- Tsai, L.H.; Delalle, I.; Caviness, V.S., Jr.; Chae, T.; Harlow, E. p35 is a neural-specific regulatory subunit of cyclin-dependent kinase 5. Nature 1994, 371, 419–423. [Google Scholar] [CrossRef]
- Chae, T.; Kwon, Y.T.; Bronson, R.; Dikkes, P.; Li, E.; Tsai, L.H. Mice lacking p35, a neuronal specific activator of cdk5, display cortical lamination defects, seizures, and adult lethality. Neuron 1997, 18, 29–42. [Google Scholar] [CrossRef] [Green Version]
- El Fatimy, R.; Miozzo, F.; Le Mouël, A.; Abane, R.; Schwendimann, L.; Sabéran-Djoneidi, D.; de Thonel, A.; Massaoudi, I.; Paslaru, L.; Hashimoto-Torii, K.; et al. Heat shock factor 2 is a stress-responsive mediator of neuronal migration defects in models of fetal alcohol syndrome. EMBO Mol. Med. 2014, 6, 1043–1061. [Google Scholar] [CrossRef]
- Fujimoto, M.; Izu, H.; Seki, K.; Fukuda, K.; Nishida, T.; Yamada, S.; Kato, K.; Yonemura, S.; Inouye, S.; Nakai, A. HSF4 is required for normal cell growth and differentiation during mouse lens development. EMBO J. 2004, 23, 4297–4306. [Google Scholar] [CrossRef] [Green Version]
- Somasundaram, T.; Bhat, S.P. Developmentally dictated expression of heat shock factors: Exclusive expression of HSF4 in the postnatal lens and its specific interaction with alphaB-crystallin heat shock promoter. J. Biol. Chem. 2004, 279, 44497–44503. [Google Scholar] [CrossRef] [Green Version]
- Shi, X.; Cui, B.; Wang, Z.; Weng, L.; Xu, Z.; Ma, J.; Xu, G.; Kong, X.; Hu, L. Removal of Hsf4 leads to cataract development in mice through down-regulation of y-crystallin and Bfsp expression. BMC Mol. Biol. 2009, 10, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roskamp, K.W.; Kozlyuk, N.; Sengupta, S.; Bierma, J.C.; Martin, R.W. Divalent cations and the divergence of βγ-crystallin function. Biochemistry 2019, 58, 4505–4518. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Li, D.; Huang, Y.; Cui, X.; Liao, S.; Wang, J.; Liu, F.; Li, C.; Gao, M.; Chen, J.; et al. HSF4 promotes G1/S arrest in human lens epithelial cells by stabilizing p53. Biochim Biophys. Acta 2015, 1853, 1808–1817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bu, L.; Jin, Y.; Shi, Y.; Chu, R.; Ban, A.; Eiberg, H.; Andres, L.; Jiang, H.; Zheng, G.; Qian, M.; et al. Mutant DNA-binding domain of HSF4 is associated with autosomal dominant lamellar and marner cataract. Nat. Genet. 2002, 31, 276–278. [Google Scholar] [CrossRef]
- Lv, H.; Huang, C.; Zhang, J.; Liu, Z.; Zhang, Z.; Xu, H.; You, Y.; Hu, J.; Li, X.; Wang, W. A novel HSF4 gene mutation causes autosomal-dominant cataracts in a Chinese family. G3 2014, 4, 823–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berry, V.; Pontikos, N.; Moore, A.; Ionides, A.C.W.; Plagnol, V.; Cheetham, M.E.; Michaelides, M. A novel missense mutation in HSF4 causes autosomal-dominant congenital lamellar cataract in a British family. Eye 2018, 32, 806–812. [Google Scholar] [CrossRef]
- Metcalf, D. The unsolved enigmas of leukemia inhibitory factor. Stem Cells 2003, 21, 5–14. [Google Scholar] [CrossRef]
- Wu, J.; Liu, T.; Rios, Z.; Mei, Q.; Lin, X.; Cao, S. Heat shock proteins and cancer. Trends Biochem. Sci. 2017, 38, 226–256. [Google Scholar] [CrossRef]
- Dai, C.; Whitesell, L.; Rogers, A.B.; Lindquist, S. Heat shock factor 1 is a powerful multifaceted modifier of carcinogenesis. Cell 2007, 130, 1005–1018. [Google Scholar] [CrossRef] [Green Version]
- Min, J.N.; Huang, L.; Zimonjic, D.B.; Moskophidis, D.; Mivechi, N.F. Selective suppression of lymphomas by functional loss of Hsf1 in a p53-deficient mouse model for spontaneous tumors. Oncogene 2007, 26, 5086–5097. [Google Scholar] [CrossRef] [Green Version]
- Jin, X.; Moskophidis, D.; Mivechi, N.F. Heat shock transcription factor 1 is a key determinant of HCC development by regulating hepatic steatosis and metabolic syndrome. Cell Metab. 2011, 14, 91–103. [Google Scholar] [CrossRef] [Green Version]
- Xi, C.; Hu, Y.; Buckhaults, P.; Moskophidis, D.; Mivechi, N.F. Heat shock factor Hsf1 cooperates with ErbB2 (Her2/Neu) protein to promote mammary tumorigenesis and metastasis. J. Biol. Chem. 2012, 287, 35646–35657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, X.; Eroglu, B.; Cho, W.; Yamaguchi, Y.; Moskophidis, D.; Mivechi, N.F. Inactivation of heat shock factor Hsf4 induces cellular senescence and suppresses tumorigenesis in vivo. Mol. Cancer Res. 2012, 10, 523–534. [Google Scholar] [CrossRef] [Green Version]
- Meng, L.; Gabai, V.L.; Sherman, M.Y. Heat-shock transcription factor HSF1 has a critical role in human epidermal growth factor receptor-2-induced cellular transformation and tumorigenesis. Oncogene 2010, 29, 5204–5213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandhapudi, S.K.; Murapa, P.; Threlkeld, Z.D.; Ward, M.; Sarge, K.D.; Snow, C.; Woodward, J.G. Heat shock transcription factor 1 is activated as a consequence of lymphocyte activation and regulates a major proteostasis network in T cells critical for cell division during stress. J. Immunol. 2013, 191, 4068–4079. [Google Scholar] [CrossRef] [PubMed]
- Dong, B.; Jaeger, A.M.; Thiele, D.J. Inhibiting heat shock factor 1 in cancer: A unique therapeutic opportunity. Trends Pharmacol. Sci. 2019, 40, 986–1005. [Google Scholar] [CrossRef] [PubMed]
- Brusselaers, N.; Ekwall, K.; Durand-Dubief, M. Copy number of 8q24.3 drives HSF1 expression and patient outcome in cancer: An individual patient data meta-analysis. Hum. Genomics 2019, 13, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Björk, J.K.; Åkerfelt, M.; Joutsen, J.; Puustinen, M.C.; Cheng, F.; Sistonen, L.; Nees, M. Heat-shock factor 2 is a suppressor of prostate cancer invasion. Oncogene 2016, 35, 1770–1784. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.N.; Ning, Z.Y.; Wang, L.; Yan, X.; Meng, Z.Q. HSF2 regulates aerobic glycolysis by suppression of FBP1 in hepatocellular carcinoma. Am. J. Cancer Res. 2019, 9, 1607–1621. [Google Scholar]
- Ma, P.; Tang, W.G.; Hu, J.W.; Hao, Y.; Xiong, L.K.; Wang, M.M.; Liu, H.; Bo, W.H.; Yu, K.H. HSP4 triggers epithelial-mesenchymal transition and promotes motility capacities of hepatocellular carcinoma cells via activating AKT. Liver Int. 2020, 40, 1211–1223. [Google Scholar] [CrossRef]
- Mustafa, D.A.; Sieuwerts, A.M.; Zheng, P.P.; Kros, J.M. Overexpression of colligin 2 in glioma vasculature is associated with overexpression of heat shock factor 2. Gene Regul. Syst. Bio. 2010, 4, 103–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, Y.H.; Cheng, H.Z.; Peng, H.; Tang, S.C.; Wang, P. Heat shock factor 2 is associated with the occurrence of lung cancer by enhancing the expression of heat shock proteins. Oncol. Lett. 2016, 12, 5106–5112. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Chen, X.; Lu, P.; Ma, W.; Yue, D.; Song, L.; Fan, Q. miR-202 promotes cell apoptosis in esophageal squamous cell carcinoma by targeting HSF2. Oncol. Res. 2017, 25, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Jin, L.; Zhang, J.; Wang, J.; Zhao, X.; Wu, G.; Yao, H.; Zhang, Z. High HSF4 expression is an independent indicator of poor overall survival and recurrence free survival in patients with primary colorectal cancer. IUBMB Life 2017, 69, 956–961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vihervaara, A.; Mahat, D.B.; Guertin, M.J.; Chu, T.; Danko, C.G.; Lis, J.T.; Sistonen, L. Transcriptional response to stress is pre-wired by promoter and enhancer architecture. Nat. Commun. 2017, 8, 255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, Y.; Xue, Y.; Zhang, L.; Feng, X.; Liu, W.; Zhang, G. Higher heat shock factor 1 expression in tumor stroma predicts poor prognosis in esophageal squamous cell carcinoma patients. J. Transl. Med. 2015, 13, 338. [Google Scholar] [CrossRef] [Green Version]
- Kourtis, N.; Lazaris, C.; Hockemeyer, K.; Balandrán, J.C.; Jimenez, A.R.; Mullenders, J.; Gong, Y.; Trimarchi, T.; Bhatt, K.; Hu, H.; et al. Oncogenic hijacking of the stress response machinery in T cell acute lymphoblastic leukemia. Nat. Med. 2018, 24, 1157–1166. [Google Scholar] [CrossRef]
- Scherz-Shouval, R.; Santagata, S.; Mendillo, M.L.; Sholl, L.M.; Ben-Aharon, I.; Beck, A.H.; Dias-Santagata, D.; Koeva, M.; Stemmer, S.M.; Whitesell, L.; et al. The reprogramming of tumor stroma by HSF1 is a potent enabler of malignancy. Cell 2014, 158, 564–578. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.Q.; Williams, H.; Prince, T.L.; Ho, E.S. Overexpressed HSF1 cancer signature genes cluster in human chromosome 8q. Hum. Genomics 2017, 11, 35. [Google Scholar] [CrossRef] [Green Version]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [Green Version]
- Joutsen, J.; Da Silva, A.J.; Luoto, J.C.; Budzynski, M.A.; Nylund, A.S.; de Thonel, A.; Concordet, J.P.; Mezger, V.; Sabéran-Djoneidi, D.; Henriksson, E.; et al. Heat shock factor 2 protects against proteotoxicity by maintaining cell–cell adhesion. Cell Rep. 2020, 30, 583–597. [Google Scholar] [CrossRef] [PubMed]
- Santagata, S.; Hu, R.; Lin, N.U.; Mendillo, M.L.; Collins, L.C.; Hankinson, S.E.; Schnitt, S.J.; Whitesell, L.; Tamimi, R.M.; Lindquist, S.; et al. High levels of nuclear heat-shock factor 1 (HSF1) are associated with poor prognosis in breast cancer. Proc. Natl. Acad. Sci. USA 2011, 108, 18378–18383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asano, Y.; Kawase, T.; Okabe, A.; Tsutsumi, S.; Ichikawa, H.; Tatebe, S.; Kitabayashi, I.; Tashiro, F.; Namiki, H.; Kondo, T.; et al. IER5 generates a novel hypo-phosphorylated active form of HSF1 and contributes to tumorigenesis. Sci. Rep. 2016, 6, 19174. [Google Scholar] [CrossRef] [PubMed]
- Vydra, N.; Janus, P.; Toma-Jonik, A.; Stokowy, T.; Mrowiec, K.; Korfanty, J.; Długajczyk, A.; Wojtaś, B.; Gielniewski, B.; Widłak, W. 17β-estradiol activates HSF1 via MAPK signaling in ERα-positive breast cancer cells. Cancers 2019, 11, 1533. [Google Scholar] [CrossRef] [Green Version]
- Yasuda, K.; Hirohashi, Y.; Mariya, T.; Murai, A.; Tabuchi, Y.; Kuroda, T.; Kusumoto, H.; Takaya, A.; Yamamoto, E.; Kubo, T.; et al. Phosphorylation of HSF1 at serine 326 residue is related to the maintenance of gynecologic cancer stem cells through expression of HSP27. Oncotarget 2017, 8, 31540–31553. [Google Scholar] [CrossRef] [Green Version]
- Whitesell, L.; Lindquist, S.L. HSP90 and the chaperoning of cancer. Nat. Rev. Cancer 2005, 5, 761–772. [Google Scholar] [CrossRef]
- Yarden, Y.; Pines, G. The ERBB network: At last, cancer therapy meets systems biology. Nat. Rev. Cancer 2012, 12, 553–563. [Google Scholar] [CrossRef]
- Schulz, R.; Streller, F.; Scheel, A.H.; Rüschoff, J.; Reinert, M.C.; Dobbelstein, M.; Marchenko, N.D.; Moll, U.M. HER2/ErbB2 activates HSF1 and thereby controls HSP90 clients including MIF in HER2-overexpressing breast cancer. Cell Death Dis. 2014, 5, e980. [Google Scholar] [CrossRef] [Green Version]
- Carpenter, R.L.; Paw, I.; Dewhirst, M.W.; Lo, H.W. Akt phosphorylates and activates HSF-1 independent of heat shock, leading to Slug overexpression and epithelial mesenchymal transition (EMT) of HER2-overexpressing breast cancer cells. Oncogene 2015, 34, 546–557. [Google Scholar] [CrossRef] [Green Version]
- Yudushkin, I. Control of Akt activity and substrate phosphorylation in cells. IUBMB Life 2020. [Google Scholar] [CrossRef] [Green Version]
- Citri, A.; Kochupurakkal, B.S.; Yarden, Y. The achilles heel of ErbB-2/HER2: Regulation by the Hsp90 chaperone machine and potential for pharmacological intervention. Cell Cycle 2004, 3, 51–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Y.J.; Pan, W.W.; Liu, S.B.; Shen, Z.F.; Xu, Y.; Hu, L.L. ERK/MAPK signalling pathway and tumorigenesis. Exp. Ther. Med. 2020, 19, 1997–2007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yallowitz, A.; Ghaleb, A.; Garcia, L.; Alexandrova, E.M.; Marchenko, N. Heat shock factor 1 confers resistance to lapatinib in ERBB2-positive breast cancer cells. Cell Death Dis. 2018, 9, 621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujimoto, M.; Takii, R.; Takaki, E.; Katiyar, A.; Nakato, R.; Shirahige, K.; Nakai, A. The HSF1-PARP13-PARP1 complex facilitates DNA repair and promotes mammary tumorigenesis. Nat. Commun. 2017, 8, 1638. [Google Scholar] [CrossRef] [PubMed]
- Chou, S.D.; Murshid, A.; Eguchi, T.; Gong, J.; Calderwood, S.K. HSF1 regulation of β-catenin in mammary cancer cells through control of HuR/elavL1 expression. Oncogene 2015, 34, 2178–2188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shang, S.; Hua, F.; Hu, Z.W. The regulation of β-catenin activity and function in cancer: Therapeutic opportunities. Oncotarget 2017, 8, 33972–33989. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Sheng, C.; Huang, L.; Zhang, H.; Huang, L.; Cheng, Z.; Zhu, Q. MiR-183/-96/-182 cluster is up-regulated in most breast cancers and increases cell proliferation and migration. Breast Cancer Res. 2014, 16, 473. [Google Scholar] [CrossRef] [Green Version]
- Sansregret, L.; Swanton, C. The role of aneuploidy in cancer evolution. CSH Perspect Med. 2017, 7, a028373. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Zhou, Y.; Xiong, X.; Huang, M.; Ying, X.; Wang, M. ALG3 is activated by heat shock factor 2 and promotes breast cancer growth. Med. Sci. Monit. 2018, 24, 3479–3487. [Google Scholar] [CrossRef]
- Chen, R.; Liliental, J.E.; Kowalski, P.E.; Lu, Q.; Cohen, S.N. Regulation of transcription of hypoxia-inducible factor-1α (HIF-1α) by heat shock factors HSF2 and HSF4. Oncogene 2011, 30, 2570–2580. [Google Scholar] [CrossRef] [Green Version]
- Forsythe, J.A.; Jiang, B.H.; Iyer, N.V.; Agani, F.; Leung, S.W.; Koos, R.D.; Semenza, G.L. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol. Cell. Biol. 1996, 16, 4604–4613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabai, V.L.; Meng, L.; Kim, G.; Mills, T.A.; Benjamin, I.J.; Sherman, M.Y. Heat shock transcription factor Hsf1 is involved in tumor progression via regulation of hypoxia-inducible factor 1 and RNA-binding protein HuR. Mol. Cell. Biol. 2012, 32, 929–940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Ma, W.; Fei, T.; Lou, Q.; Zhang, Y.; Cui, X.; Qin, X.; Zhang, J.; Liu, G.; Dong, Z.; et al. Upregulation of heat shock factor 1 transcription activity is associated with hepatocellular carcinoma progression. Mol. Med. Rep. 2014, 10, 2313–2321. [Google Scholar] [CrossRef] [Green Version]
- Ma, W.; Zhang, Y.; Mu, H.; Qing, X.; Li, S.; Cui, X.; Lou, Q.; Ma, Y.; Pu, H.; Hu, Y. Glucose regulates heat shock factor 1 transcription activity via mTOR pathway in HCC cell lines. Cell Biol. Int. 2015, 39, 1217–1224. [Google Scholar] [CrossRef] [PubMed]
- Cigliano, A.; Wang, C.; Pilo, M.G.; Szydlowska, M.; Brozzetti, S.; Latte, G.; Pes, G.M.; Pascale, R.M.; Seddaiu, M.A.; Vidili, G.; et al. Inhibition of HSF1 suppresses the growth of hepatocarcinoma cell lines in vitro and AKT-driven hepatocarcinogenesis in mice. Oncotarget 2017, 8, 54149–54159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, J.C.; Thomas, D.M.; Choong, P.F.; Dass, C.R. RECK-a newly discovered inhibitor of metastasis with prognostic significance in multiple forms of cancer. Cancer Metastasis Rev. 2007, 26, 675–683. [Google Scholar] [CrossRef]
- Li, Y.; Xu, D.; Bao, C.; Zhang, Y.; Chen, D.; Zhao, F.; Ding, J.; Liang, L.; Wang, Q.; Liu, L.; et al. MicroRNA-135b, a HSF1 target, promotes tumor invasion and metastasis by regulating RECK and EVI5 in hepatocellular carcinoma. Oncotarget 2015, 6, 2421–2433. [Google Scholar] [CrossRef] [Green Version]
- Jiang, B. Aerobic glycolysis and high level of lactate in cancer metabolism and microenvironment. Genes Dis. 2017, 4, 25–27. [Google Scholar] [CrossRef]
- Schödel, J.; Oikonomopoulos, S.; Ragoussis, J.; Pugh, C.W.; Ratcliffe, P.J.; Mole, D.R. High-resolution genome-wide mapping of HIF-binding sites by ChIP-seq. Blood 2011, 117, e207–e217. [Google Scholar] [CrossRef] [Green Version]
- Shi, L.; He, C.; Li, Z.; Wang, Z.; Zhang, Q. FBP1 modulates cell metabolism of breast cancer cells by inhibiting the expression of HIF-1alpha. Neoplasma 2017, 64, 535–542. [Google Scholar] [CrossRef]
- Wang, Y.; Theriault, J.R.; He, H.; Gong, J.; Calderwood, S.K. Expression of a dominant negative heat shock factor-1 construct inhibits aneuploidy in prostate carcinoma cells. J. Biol. Chem. 2004, 279, 32651–32659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Bokhoven, A.; Varella-Garcia, M.; Korch, C.; Johannes, W.U.; Smith, E.E.; Miller, H.L.; Nordeen, S.K.; Miller, G.J.; Lucia, M.S. Molecular characterization of human prostate carcinoma cell lines. Prostate 2003, 57, 205–225. [Google Scholar] [CrossRef] [PubMed]
- Davoli, T.; de Lange, T. The causes and consequences of polyploidy in normal development and cancer. Annu. Rev. Cell Dev. Biol. 2011, 27, 585–610. [Google Scholar] [CrossRef] [Green Version]
- Björk, J.K.; Ahonen, I.; Mirtti, T.; Erickson, A.; Rannikko, A.; Bützow, A.; Nordling, S.; Lundin, J.; Lundin, M.; Sistonen, L.; et al. Increased HSF1 expression predicts shorter disease-specific survival of prostate cancer patients following radical prostatectomy. Oncotarget 2018, 9, 31200–31213. [Google Scholar] [CrossRef] [Green Version]
- Cui, J.; Tian, H.; Chen, G. Upregulation of nuclear heat shock factor 1 contributes to tumor angiogenesis and poor survival in patients with non-small cell lung cancer. Ann. Thorac. Surg. 2015, 100, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Ocejo-Garcia, M.; Baokbah, T.A.; Ashurst, H.L.; Cowlishaw, D.; Soomro, I.; Coulson, J.M.; Woll, P.J. Roles for USF-2 in lung cancer proliferation and bronchial carcinogenesis. J. Pathol. 2005, 206, 151–159. [Google Scholar] [CrossRef]
- Lee, S.S.; Kwon, S.H.; Sung, J.S.; Han, M.Y.; Park, Y.M. Cloning and characterization of the rat Hsf2 promoter: A critical role of proximal E-box element and USF protein in Hsf2 regulation in different compartments of the brain. Biochim. Biophys. Acta 2003, 1625, 52–63. [Google Scholar] [CrossRef]
- Tsukao, Y.; Yamasaki, M.; Miyazaki, Y.; Makino, T.; Takahashi, T.; Kurokawa, Y.; Miyata, H.; Nakajima, K.; Takiguchi, S.; Mimori, K.; et al. Overexpression of heat-shock factor 1 is associated with a poor prognosis in esophageal squamous cell carcinoma. Oncol. Lett. 2017, 3, 1819–1825. [Google Scholar] [CrossRef]
- Kawanishi, K.; Shiozaki, H.; Doki, Y.; Sakita, I.; Inoue, M.; Yano, M.; Tsujinaka, T.; Shamma, A.; Monden, M. Prognostic significance of heat shock proteins 27 and 70 in patients with squamous cell carcinoma of the esophagus. Cancer 1999, 85, 1649–1657. [Google Scholar] [CrossRef]
- Noguchi, T.; Takeno, S.; Shibata, T.; Uchida, Y.; Yokoyama, S.; Müller, W. Expression of heat shock protein 70 in grossly resected esophageal squamous cell carcinoma. Ann. Thorac. Surg. 2002, 74, 222–226. [Google Scholar] [CrossRef]
- Li, J.; Song, P.; Jiang, T.; Dai, D.; Wang, H.; Sun, J.; Zhu, L.; Xu, W.; Feng, L.; Shin, V.Y.; et al. Heat shock factor 1 epigenetically stimulates glutaminase-1-dependent mTOR activation to promote colorectal carcinogenesis. Mol. Ther. 2018, 26, 1828–1839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Song, P.; Zhu, L.; Aziz, N.; Zhou, Q.; Zhang, Y.; Xu, W.; Feng, L.; Chen, D.; Wang, X.; et al. Synthetic lethality of glutaminolysis inhibition, autophagy inactivation and asparagine depletion in colon cancer. Oncotarget 2017, 8, 42664–42672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobs, A.T.; Marnett, L.J. HSF1-mediated BAG3 expression attenuates apoptosis in 4-hydroxynonenal-treated colon cancer cells via stabilization of anti-apoptotic Bcl-2 proteins. J. Biol. Chem. 2009, 284, 9176–9183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.; Mivechi, N. Association and regulation of heat shock transcription factor 4b with both extracellular signal-regulated kinase mitogen-activated protein kinase and dual-specificity tyrosine phosphatase DUSP26. Mol. Cell. Biol. 2006, 26, 3282–3294. [Google Scholar] [CrossRef] [Green Version]
- Lü, S.; Wang, J. The resistance mechanisms of proteasome inhibitor bortezomib. Biomark Res. 2013, 1, 13. [Google Scholar] [CrossRef] [Green Version]
- Damaskos, C.; Garmpis, N.; Valsami, S.; Kontos, M.; Spartalis, E.; Kalampokas, T.; Kalampokas, E.; Athanasiou, A.; Moris, D.; Daskalopoulou, A.; et al. Histone deacetylase inhibitors: An attractive therapeutic strategy against breast cancer. Anticancer Res. 2017, 37, 35–46. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| HSF | Cancer Type | Expression in Cells and Tissues | Effect on Tumorigenesis |
|---|---|---|---|
| HSF1 | HCC | High | Promotes cell proliferation, growth, migration, invasion, and survival as well as kinase function, lipid metabolism, and glycolysis. |
| HSF2 | HCC | High | Promotes cell proliferation and aerobic glycolysis. |
| HSF4 | HCC | High | Promotes cell proliferation, migration, kinase function, and EMT. |
| HSF1 | Breast cancer | High | Promotes cell motility, metastasis, and survival as well as receptor and kinase maturation, stemness, drug resistance, DNA repair, and EMT. |
| HSF2 | Breast cancer | High | Promotes cell proliferation, migration, and aneuploidy. |
| HSF1 | Prostate cancer | High | Promotes development of polyploidy, high Gleason score, and cancer re-occurrence. |
| HSF2 | Prostate cancer | Low | Promotes organoid differentiation, invasive growth, and high Gleason score. |
| HSF1 | Lung Cancer | High | Decreases patient survival. Promotes angiogenesis and metastasis. |
| HSF2 | Lung Cancer | High | Promotes cell proliferation, migration, and expression of HSPs. |
| HSF1 | ESCC | High | Promotes cell survival and expression of HSPs. |
| HSF2 | ESCC | High | Promotes cell survival and expression of HSPs. |
| HSF1 | CRC | High | Promotes expression of anti-apoptotic proteins, cell growth, and glutaminolysis. |
| HSF4 | CRC | High | Decreases patient survival. Promotes cancer re-ocurrence. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Puustinen, M.C.; Sistonen, L. Molecular Mechanisms of Heat Shock Factors in Cancer. Cells 2020, 9, 1202. https://doi.org/10.3390/cells9051202
Puustinen MC, Sistonen L. Molecular Mechanisms of Heat Shock Factors in Cancer. Cells. 2020; 9(5):1202. https://doi.org/10.3390/cells9051202
Chicago/Turabian StylePuustinen, Mikael Christer, and Lea Sistonen. 2020. "Molecular Mechanisms of Heat Shock Factors in Cancer" Cells 9, no. 5: 1202. https://doi.org/10.3390/cells9051202
APA StylePuustinen, M. C., & Sistonen, L. (2020). Molecular Mechanisms of Heat Shock Factors in Cancer. Cells, 9(5), 1202. https://doi.org/10.3390/cells9051202