P38 MAPK Promotes Migration and Metastatic Activity of BRAF Mutant Melanoma Cells by Inducing Degradation of PMCA4b

 , ,
, ,  , ,
, ,  and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Human Cell Lines
2.1.1. Cell Culture
2.1.2. Cell Treatments, Chemical Reagents
2.1.3. Generation of Stable Cell Lines
2.1.4. Transient Cell Transfection
2.2. Cell Treatments and Measurements
2.2.1. Quantitative Real-Time Polymerase Chain Reaction
2.2.2. Western Blot Analysis
2.2.3. Immunofluorescence Microscopy
2.2.4. Ca2+ Signal Measurements
2.2.5. Transmission Electron Microscopy (TEM)
2.2.6. Sulforhodamine B (SRB) Assay
2.2.7. Cell Cycle Analysis
2.2.8. Colony-Forming Assay
2.2.9. siRNA Transfection
2.3. In vitro Cell Function Assays
2.3.1. Cell Migration Assay
2.3.2. Spheroid-Forming Assay
2.3.3. Reversal of Multicellular Spheroid (MCS) Formation
2.4. Statistical Analysis
3. Results
3.1. Inhibition of p38 MAPK—But Not JNK or NF-kB—Upregulates PMCA4b in BRAF Mutant Melanoma Cells
3.2. Inhibition of p38 MAPK Increased Stability and Plasma Membrane Abundance of PMCA4b Resulting in Enhanced Ca2+ Clearance
3.3. PMCA4b Is Degraded through the Endo/Lysosomal System In a p38 MAPK Dependent Manner
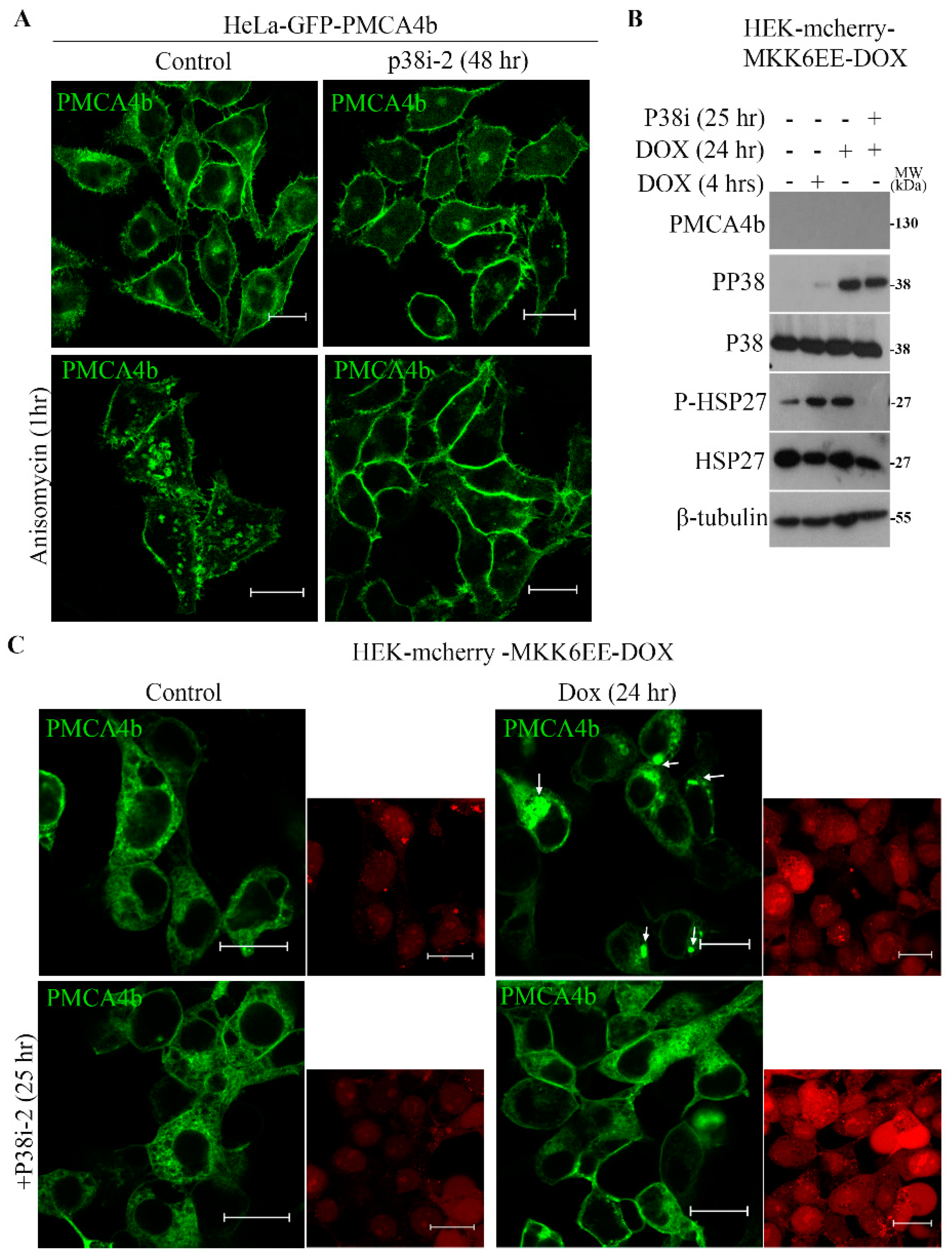
3.4. P38 MAPK is a Modulator of PMCA4b Trafficking
3.5. The P38 Inhibitor Induced Cell Cycle Arrest and Reduced Colony Formation but Displayed Low Cytotoxicity
3.6. P38 Inhibitor Decreased Migratory Activity of A375 Melanoma Cells through PMCA4b
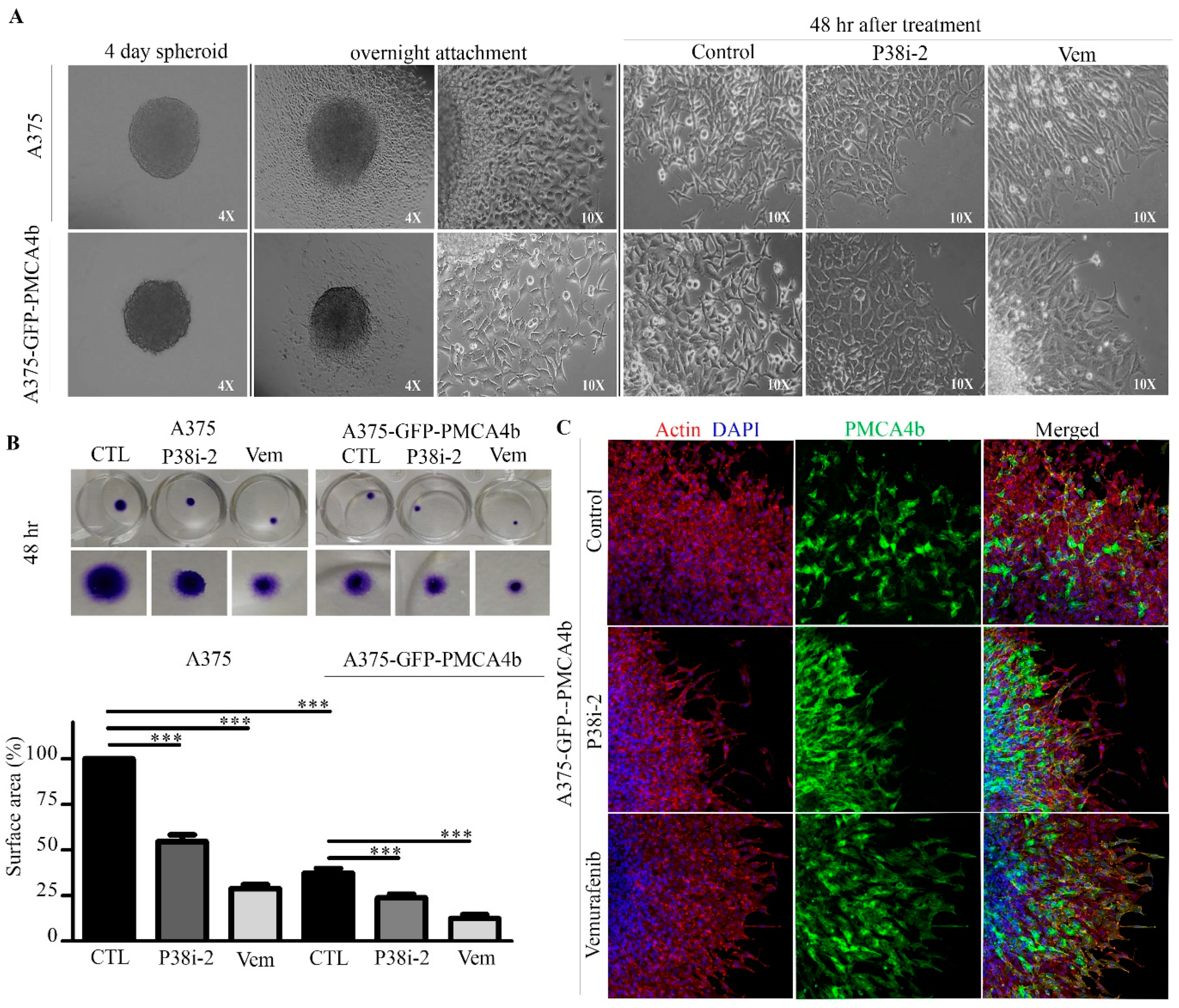
3.7. PMCA4b and p38 Inhibitor Moderately Reduce Spheroid Growth
3.8. P38 Inhibitor Reduced Metastatic Activity of A375 Cells In Vitro
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Narayanan, D.L.; Saladi, R.N.; Fox, J.L. Ultraviolet radiation and skin cancer. Int. J. Dermatol. 2010, 49, 978–986. [Google Scholar] [CrossRef] [PubMed]
- Ascierto, P.A.; Kirkwood, J.M.; Grob, J.J.; Simeone, E.; Grimaldi, A.M.; Maio, M.; Palmieri, G.; Testori, A.; Marincola, F.M.; Mozzillo, N. The role of BRAF V600 mutation in melanoma. J. Transl. Med. 2012, 10, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maurer, G.; Tarkowski, B.; Baccarini, M. Raf kinases in cancer-roles and therapeutic opportunities. Oncogene 2011, 30, 3477–3488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estrada, Y.; Dong, J.; Ossowski, L. Positive crosstalk between ERK and p38 in melanoma stimulates migration and in vivo proliferation. Pigment Cell Melanoma Res. 2009, 22, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Bergami, P.; Huang, C.; Goydos, J.S.; Yip, D.; Bar-Eli, M.; Herlyn, M.; Smalley, K.S.; Mahale, A.; Eroshkin, A.; Aaronson, S.; et al. Rewired ERK-JNK signaling pathways in melanoma. Cancer Cell 2007, 11, 447–460. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.; DeGuzman, A.; Bucana, C.D.; Fidler, I.J. Nuclear factor-kappaB activity correlates with growth, angiogenesis, and metastasis of human melanoma cells in nude mice. Clin. Cancer Res. 2000, 6, 2573–2581. [Google Scholar]
- Marchi, S.; Pinton, P. Alterations of calcium homeostasis in cancer cells. Curr. Opin. Pharmacol. 2016, 29, 1–6. [Google Scholar] [CrossRef]
- Berridge, M.J.; Bootman, M.D.; Roderick, H.L. Calcium signalling: Dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 2003, 4, 517–529. [Google Scholar] [CrossRef] [Green Version]
- Bultynck, G.; Campanella, M. Tumor suppressive Ca2+ signaling is driven by IP3 receptor fitness. Cell Stress 2017, 1, 73–78. [Google Scholar] [CrossRef]
- Bong, A.H.L.; Monteith, G.R. Calcium signaling and the therapeutic targeting of cancer cells. Biochim Biophys Acta Mol Cell Res. 2018, 1865, 1786–1794. [Google Scholar] [CrossRef]
- Prevarskaya, N.; Skryma, R.; Shuba, Y. Ion channels and the hallmarks of cancer. Trends Mol. Med. 2010, 16, 107–121. [Google Scholar] [CrossRef] [PubMed]
- Monteith, G.; Davis, F.; Roberts-Thomson, S. Calcium channels and pumps in cancer: Changes and consequences. J. Biol. Chem. 2012, 287, 31666–31673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krebs, J. The plethora of PMCA isoforms: Alternative splicing and differential expression. Biochim. Biophys. Acta 2015, 1853, 2018–2024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padányi, R.; Pászty, K.; Hegedűs, L.; Varga, K.; Papp, B.; Penniston, J.T.; Enyedi, Á. Multifaceted plasma membrane Ca(2+) pumps: From structure to intracellular Ca(2+) handling and cancer. Biochim. Biophys. Acta 2016, 1863, 1351–1363. [Google Scholar] [CrossRef] [Green Version]
- Roberts-Thomson, S.; Curry, M.; Monteith, G. Plasma membrane calcium pumps and their emerging roles in cancer. World J. Biol. Chem. 2010, 1, 248–253. [Google Scholar] [CrossRef]
- Macià, A.; Herreros, J.; Martí, R.M.; Cantí, C. Calcium channel expression and applicability as targeted therapies in melanoma. Biomed. Res. Int. 2015, 2015, 587135. [Google Scholar] [CrossRef]
- Hegedũs, L.; Garay, T.; Molnárm, E.; Varga, K.; Bilecz, Á.; Törökm, S.; Padányi, R.; Pászty, K.; Wolf, M.; Grusch, M.; et al. The plasma membrane CA2+ pump PMCA4b inhibits the migratory and metastatic activity of BRAF mutant melanoma cells. Int. J. Cancer 2017, 140, 2758–2770. [Google Scholar] [CrossRef] [Green Version]
- Varga, K.; Pászty, K.; Padányi, R.; Hegedűs, L.; Brouland, J.P.; Papp, B.; Enyedi, A. Histone deacetylase inhibitor- and PMA induced upregulation of PMCA4b enhances Ca2+ clearance from MCF-7 breast cancer cells. Cell Calcium 2014, 55, 78–92. [Google Scholar] [CrossRef]
- Varga, K.; Hollósi, A.; Pászty, K.; Hegedűs, L.; Szakács, G.; Tímár, J.; Papp, B.; Enyedi, Á.; Padányi, R. Expression of calcium pumps is differentially regulated by histone deacetylase inhibitors and estrogen receptor alpha in breast cancer cells. BMC Cancer 2018, 18, 1029. [Google Scholar] [CrossRef] [Green Version]
- Rüschoff, J.H.; Brandenburger, T.; Strehler, E.E.; Filoteo, A.G.; Heinmöller, E.; Aumüller, G.; Wilhelm, B. Plasma membrane calcium ATPase expression in human colon multistep carcinogenesis. Cancer Invest. 2012, 30, 251–257. [Google Scholar] [CrossRef]
- Ribiczey, P.; Tordai, A.; Andrikovics, H.; Filoteo, A.G.; Penniston, J.T.; Enouf, J.; Enyedi, A.; Papp, B.; Kovács, T. Isoform-specific up-regulation of plasma membrane Ca2+ATPase expression during colon and gastric cancer cell differentiation. Cell Calcium 2007, 42, 590–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegedüs, L.; Padányi, R.; Molnár, J.; Pászty, K.; Varga, K.; Kenessey, I.; Sárközy, E.; Wolf, M.; Grusch, M.; Hegyi, Z.; et al. Histone deacetylase inhibitor treatment increases the expression of the plasma membrane Ca2+ pump PMCA4b and inhibits the migration of melanoma cells independent of ERK. Front. Oncol. 2017, 7, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vőfély, G.; Berecz, T.; Szabó, E.; Szebényi, K.; Hathy, E.; Orbán, T.I.; Sarkadi, B.; Homolya, L.; Marchetto, M.C.; Réthelyi, J.M.; et al. Characterization of calcium signals in human induced pluripotent stem cell-derived dentate gyrus neuronal progenitors and mature neurons, stably expressing an advanced calcium indicator protein. Mol. Cell Neurosci. 2018, 88, 222–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bach, M.; Grigat, S.; Pawlik, B.; Fork, C.; Utermöhlen, O.; Pal, S.; Banczyk, D.; Lazar, A.; Schömig, E.; Gründemann, D. Fast set-up of doxycycline-inducible protein expression in human cell lines with a single plasmid based on Epstein-Barr virus replication and the simple tetracycline repressor. FEBS J. 2007, 274, 783–790. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, D.P.; Parker, T.L.; Walker, D.A.; Alexander, C.; Ashford, M.B.; Gellert, P.R.; Garnett, M.C. Multiplexing spheroid volume, resazurin and acid phosphatase viability assays for high-throughput screening of tumour spheroids and stem cell neurospheres. PLoS ONE 2014, 9, e103817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunjithapatham, R.; Karthikeyan, S.; Geschwind, J.F.; Kieserman, E.; Lin, M.; Fu, D.X.; Ganapathy-Kanniappan, S. Reversal of anchorage-independent multicellular spheroid into a monolayer mimics a metastatic model. Sci. Rep. 2014, 4, 6816. [Google Scholar] [CrossRef]
- Corazzari, M.; Rapino, F.; Ciccosanti, F.; Giglio, P.; Antonioli, M.; Conti, B.; Fimia, G.M.; Lovat, P.E.; Piacentini, M. Oncogenic BRAF induces chronic ER stress condition resulting in increased basal autophagy and apoptotic resistance of cutaneous melanoma. Cell Death Differ. 2015, 22, 946–958. [Google Scholar] [CrossRef]
- Vergarajauregui, S.; San Miguel, A.; Puertollano, R. Activation of p38 mitogen-activated protein kinase promotes epidermal growth factor receptor internalization. Traffic 2006, 7, 686–698. [Google Scholar] [CrossRef] [Green Version]
- Costa, E.C.; Moreira, A.F.; de Melo-Diogo, D.; Gaspar, V.M.; Carvalho, M.P.; Correia, I.J. 3D tumor spheroids: An overview on the tools and techniques used for their analysis. Biotechnol. Adv. 2016, 34, 1427–1441. [Google Scholar] [CrossRef]
- Huang, R.; Rofstad, E.K. Integrins as therapeutic targets in the organ specific metastsasis of human malignant melanoma. J. Exp. Clin. Cancer Res. 2018, 37, 92. [Google Scholar] [CrossRef]
- Mariotti, A.; Kedeshian, P.A.; Dans, M.; Curatola, A.M.; Gagnoux-Palacios, L.; Giancotti, F.G. EGF-R signaling through Fyn kinase disrupts the function of integrin alpha6beta4 at hemidesmosomes: Role in epithelial cell migration and carcinoma invasion. J. Cell Biol. 2001, 155, 447–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuadrado, A.; Nebreda, A.R. Mechanisms and functions of p38 MAPK signalling. Biochem. J. 2010, 429, 403–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zarubin, T.; Han, J. Activation and signaling of the p38 MAP kinase pathway. Cell Res. 2005, 15, 11–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuenda, A.; Rousseau, S. p38 MAP-Kinases pathway regulation, function and role in human diseases. Biochim. Biophys. Acta 2007, 1773, 1358–1375. [Google Scholar] [CrossRef] [Green Version]
- Jin, E.J.; Kiral, F.R.; Hiesinger, P.R. The where, what, and when of membrane protein degradation in neurons. Dev. Neurobiol. 2018, 78, 283–297. [Google Scholar] [PubMed] [Green Version]
- Li, S.; Song, Y.; Quach, C.; Guo, H.; Jang, G.B.; Maazi, H.; Zhao, S.; Sands, N.A.; Liu, Q.; In, G.K.; et al. Transcriptional regulation of autophagy-lysosomal function in BRAF driven melanoma progression and chemoresistance. Nat. Commun. 2019, 10, 1693. [Google Scholar] [CrossRef] [Green Version]
- Pelkmans, L.; Fava, E.; Grabner, H.; Hannus, M.; Habermann, B.; Krausz, E.; Zerial, M. Genome-wide analysis of human kinases in clathrin- and caveolae/raft-mediated endocytosis. Nature 2005, 436, 78–86. [Google Scholar] [CrossRef]
- Ferrington, D.A.; Chen, X.; Krainev, A.G.; Michaelis, E.K.; Bigelow, D.J. Protein half-lives of calmodulin and the plasma membrane Ca-ATPase in rat brain. Biochem. Biophys. Res. Commun. 1997, 23, 163–165. [Google Scholar] [CrossRef]
- Antalffy, G.; Pászty, K.; Varga, K.; Hegedűs, L.; Enyedi, Á.; Padányi, R. A C-terminal di-leucine motif controls plasma membrane expression of PMCA4b. Biochim. Biophys. Acta 2013, 1833, 2561–2572. [Google Scholar] [CrossRef] [Green Version]
- Kalal, B.S.; Pai, V.R.; Upadhya, D. Valproic Acid Reduces Tumor Cell Survival and Proliferation with Inhibitors of Downstream Molecules of Epidermal Growth Factor Receptor Pathway. J. Pharmacol. Pharmacother. 2018, 9, 11–16. [Google Scholar]
- Matrone, A.; Grossi, V.; Chiacchiera, F.; Fina, E.; Cappellari, M.; Caringella, A.M.; Di Naro, E.; Loverro, G.; Simone, C. p38 alpha is required for ovarian cancer cell metabolism and survival. Int. J. Gynecol. Cancer 2010, 20, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Grossi, V.; Liuzzi, M.; Murzilli, S.; Martelli, N.; Napoli, A.; Ingravallo, G.; Del Rio, A.; Simone, C. Sorafenib inhibits p38α activity in colorectal cancer cells and synergizes with the DFG-in inhibitor SB202190 to increase apoptotic response. Cancer Biol. Ther. 2012, 13, 1471–1481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Wang, X.; Qin, X.; Wang, X.; Liu, F.; White, E.; Zheng, X.F. PP2AC level determines differential programming of p38-TSC-mTOR signaling and therapeutic response to p38-Targeted Therapy in Colorectal Cancer. EBioMedicine 2015, 2, 1944–1956. [Google Scholar] [CrossRef] [Green Version]
- Rousseau, S.; Houle, F.; Landry, J.; Huot, J. p38 MAP kinase activation by vascular endothelial growth factor mediates actin reorganization and cell migration in human endothelial cells. Oncogene 1997, 15, 2169–2177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pászty, K.; Caride, A.J.; Bajzer, Ž.; Offord, C.P.; Padányi, R.; Hegedűs, L.; Varga, K.; Strehler, E.E.; Enyedi, A. Plasma membrane Ca2+-ATPases can shape the pattern of Ca2+ transients induced by store-operated Ca2+ entry. Sci Signal. 2015, 8, ra19. [Google Scholar] [CrossRef] [Green Version]
- Tsai, F.C.; Kuo, G.H.; Chang, S.W.; Tsai, P.J. Ca2+ signaling in cytoskeletal reorganization, cell migration, and cancer metastasis. Biomed Res Int. 2015, 2015, 409245. [Google Scholar] [CrossRef] [Green Version]
- Tsai, F.C.; Seki, A.; Yang, H.W.; Hayer, A.; Carrasco, S.; Malmersjö, S.; Meyer, T. A polarized Ca2+, diacylglycerol and STIM1 signalling system regulates directed cell migration. Nat Cell Biol. 2014, 16, 133–144. [Google Scholar] [CrossRef] [Green Version]
- Dalghi, M.G.; Ferreira-Gomes, M.; Rossi, J.P. Regulation of the Plasma Membrane Calcium ATPases by the actin cytoskeleton. Biochem. Biophys. Res. Commun. 2018, 506, 347–354. [Google Scholar] [CrossRef]
- Roy, S.; Roy, S.; Kar, M.; Padhi, S.; Saha, A.; Anuja, K.; Banerjee, B. Role of p38 MAPK in disease relapse and therapeutic resistance by maintenance of cancer stem cells in head and neck squamous cell carcinoma. J. Oral Pathol. Med. 2018, 47, 492–501. [Google Scholar] [CrossRef]
- Demuth, T.; Reavie, L.B.; Rennert, J.L.; Nakada, M.; Nakada, S.; Hoelzinger, D.B.; Beaudry, C.E.; Henrichs, A.N.; Anderson, E.M.; Berens, M.E. MAP-ing glioma invasion: Mitogen activated protein kinase 3 and p38 drive glioma invasion and progression and predict patient survival. Mol. Cancer Ther. 2007, 6, 1212–1222. [Google Scholar] [CrossRef] [Green Version]
- Cui, X.; Hartanto, Y.; Zhang, H. Advances in multicellular spheroids formation. J. R. Soc. Interface 2017, 14, 20160877. [Google Scholar] [CrossRef] [PubMed]









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Naffa, R.; Vogel, L.; Hegedűs, L.; Pászty, K.; Tóth, S.; Kelemen, K.; Singh, N.; Reményi, A.; Kállay, E.; Cserepes, M.; et al. P38 MAPK Promotes Migration and Metastatic Activity of BRAF Mutant Melanoma Cells by Inducing Degradation of PMCA4b. Cells 2020, 9, 1209. https://doi.org/10.3390/cells9051209
Naffa R, Vogel L, Hegedűs L, Pászty K, Tóth S, Kelemen K, Singh N, Reményi A, Kállay E, Cserepes M, et al. P38 MAPK Promotes Migration and Metastatic Activity of BRAF Mutant Melanoma Cells by Inducing Degradation of PMCA4b. Cells. 2020; 9(5):1209. https://doi.org/10.3390/cells9051209
Chicago/Turabian StyleNaffa, Randa, Lisa Vogel, Luca Hegedűs, Katalin Pászty, Sarolta Tóth, Kornélia Kelemen, Neha Singh, Attila Reményi, Enikő Kállay, Mihály Cserepes, and et al. 2020. "P38 MAPK Promotes Migration and Metastatic Activity of BRAF Mutant Melanoma Cells by Inducing Degradation of PMCA4b" Cells 9, no. 5: 1209. https://doi.org/10.3390/cells9051209
APA StyleNaffa, R., Vogel, L., Hegedűs, L., Pászty, K., Tóth, S., Kelemen, K., Singh, N., Reményi, A., Kállay, E., Cserepes, M., Tóvári, J., Grusch, M., & Enyedi, Á. (2020). P38 MAPK Promotes Migration and Metastatic Activity of BRAF Mutant Melanoma Cells by Inducing Degradation of PMCA4b. Cells, 9(5), 1209. https://doi.org/10.3390/cells9051209