On the Host Side of the Hepatitis E Virus Life Cycle


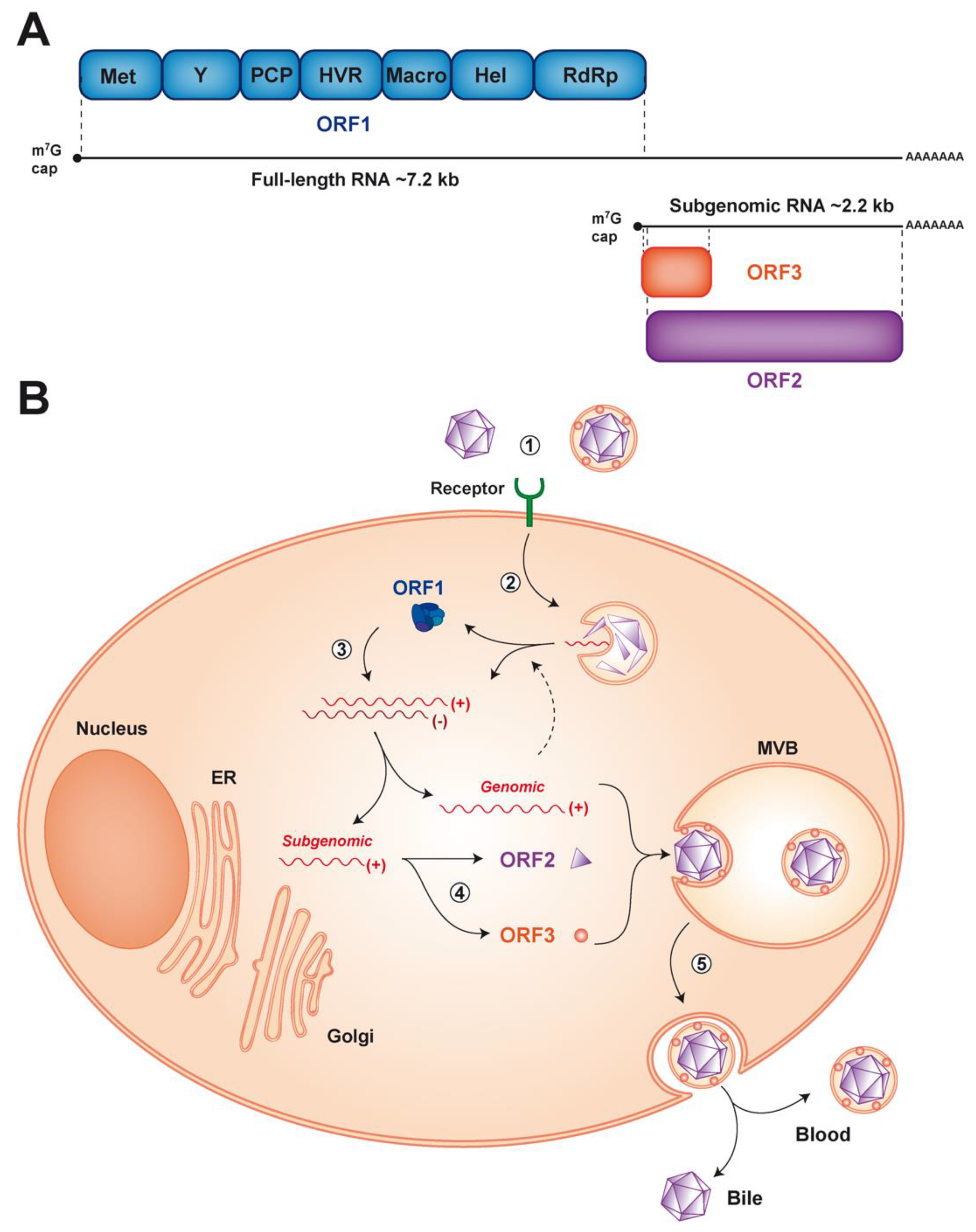{kind=link}
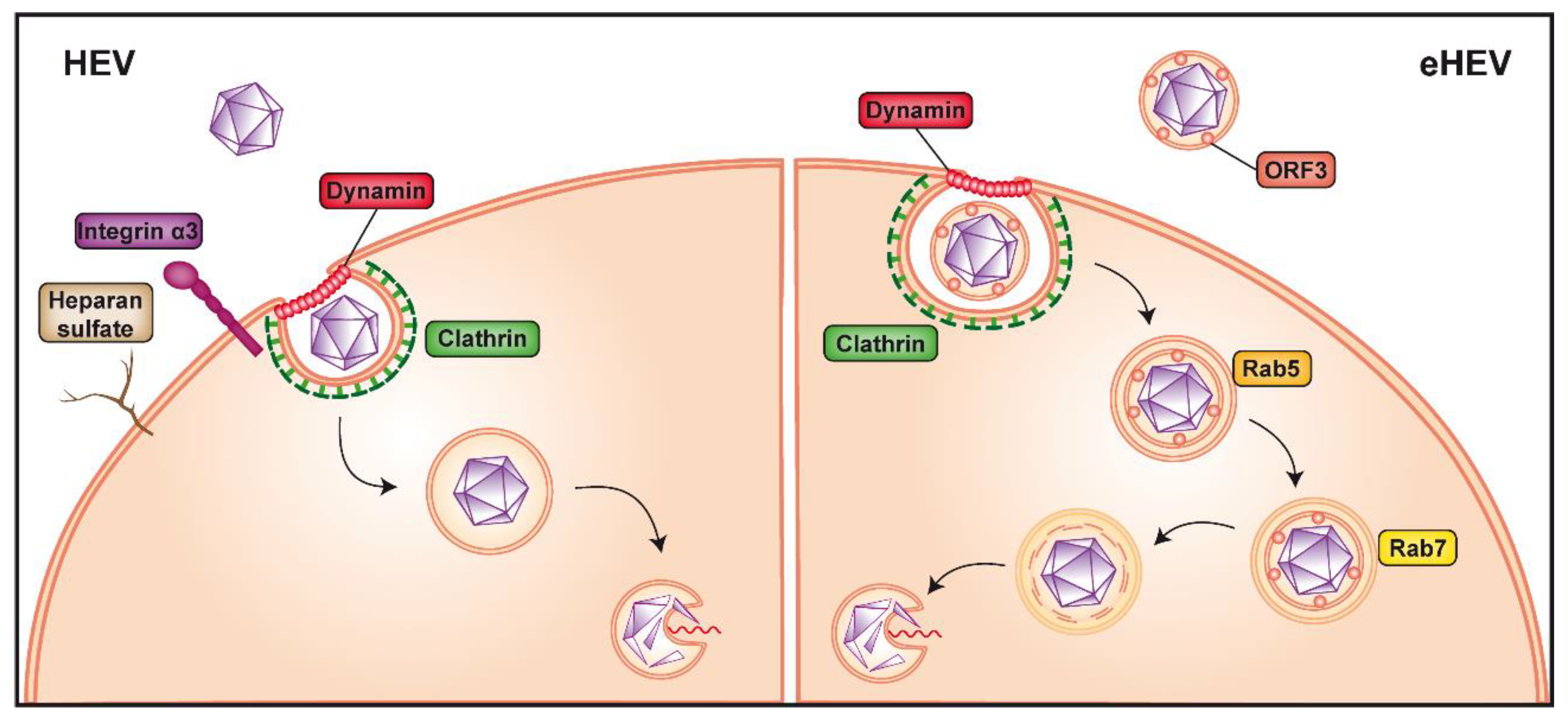{kind=link}
{kind=link}
Abstract
:1. Introduction
2. HEV Entry
3. Viral RNA Replication
4. Virion Assembly and Infectious Particle Release
5. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Khuroo, M.S. Study of an epidemic of non-A, non-B hepatitis. Possibility of another human hepatitis virus distinct from post-transfusion non-A, non-B type. Am. J. Med. 1980, 68, 818–824. [Google Scholar] [CrossRef]
- Balayan, M.S.; Andjaparidze, A.G.; Savinskaya, S.S.; Ketiladze, E.S.; Braginsky, D.M.; Savinov, A.P.; Poleschuk, V.F. Evidence for a virus in non-A, non-B hepatitis transmitted via the fecal-oral route. Intervirology 1983, 20, 23–31. [Google Scholar]
- Reyes, G.R.; Purdy, M.A.; Kim, J.P.; Luk, K.C.; Young, L.M.; Fry, K.E.; Bradley, D.W. Isolation of a cDNA from the virus responsible for enterically transmitted non-A, non-B hepatitis. Science 1990, 247, 1335–1339. [Google Scholar] [CrossRef] [PubMed]
- Tam, A.W.; Smith, M.M.; Guerra, M.E.; Huang, C.C.; Bradley, D.W.; Fry, K.E.; Reyes, G.R. Hepatitis E virus (HEV): Molecular cloning and sequencing of the full-length viral genome. Virology 1991, 185, 120–131. [Google Scholar] [CrossRef]
- Smith, D.B.; Simmonds, P.; Jameel, S.; Emerson, S.U.; Harrison, T.J.; Meng, X.J.; Okamoto, H.; Van der Poel, W.H.; Purdy, M.A. Consensus proposals for classification of the family Hepeviridae. J. Gen. Virol. 2015, 96, 1191–1192. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.H.; Tan, B.H.; Teo, E.C.; Lim, S.G.; Dan, Y.Y.; Wee, A.; Aw, P.P.; Zhu, Y.; Hibberd, M.L.; Tan, C.K.; et al. Chronic infection with camelid hepatitis E virus in a liver transplant recipient who regularly consumes camel meat and milk. Gastroenterology 2016, 150, 355.e353–357.e353. [Google Scholar] [CrossRef] [Green Version]
- Nimgaonkar, I.; Ding, Q.; Schwartz, R.E.; Ploss, A. Hepatitis E virus: Advances and challenges. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 96–110. [Google Scholar] [CrossRef]
- Abravanel, F.; Lhomme, S.; El Costa, H.; Schvartz, B.; Peron, J.M.; Kamar, N.; Izopet, J. Rabbit hepatitis E virus infections in humans, France. Emerg. Infect. Dis. 2017, 23, 1191–1193. [Google Scholar] [CrossRef] [Green Version]
- Sahli, R.; Fraga, M.; Semela, D.; Moradpour, D.; Gouttenoire, J. Rabbit HEV in immunosuppressed patients with hepatitis E acquired in Switzerland. J. Hepatol. 2019, 70, 1023–1025. [Google Scholar] [CrossRef] [Green Version]
- Sridhar, S.; Yip, C.C.Y.; Wu, S.; Cai, J.; Zhang, A.J.; Leung, K.H.; Chung, T.W.H.; Chan, J.F.W.; Chan, W.M.; Teng, J.L.L.; et al. Rat hepatitis E virus as cause of persistent hepatitis after liver transplant. Emerg. Infect. Dis. 2018, 24, 2241–2250. [Google Scholar] [CrossRef] [Green Version]
- Sridhar, S.; Yip, C.C.; Wu, S.; Chew, N.F.; Leung, K.H.; Chan, J.F.; Zhao, P.S.; Chan, W.M.; Poon, R.W.; Tsoi, H.W.; et al. Transmission of rat hepatitis E virus infection to humans in Hong Kong: A clinical and epidemiological analysis. Hepatology 2020. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Heller, B.; Capuccino, J.M.; Song, B.; Nimgaonkar, I.; Hrebikova, G.; Contreras, J.E.; Ploss, A. Hepatitis E virus ORF3 is a functional ion channel required for release of infectious particles. Proc. Natl. Acad. Sci. USA 2017, 114, 1147–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graff, J.; Torian, U.; Nguyen, H.; Emerson, S.U. A bicistronic subgenomic mRNA encodes both the ORF2 and ORF3 proteins of hepatitis E virus. J. Virol. 2006, 80, 5919–5926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ichiyama, K.; Yamada, K.; Tanaka, T.; Nagashima, S.; Jirintai; Takahashi, M.; Okamoto, H. Determination of the 5′-terminal sequence of subgenomic RNA of hepatitis E virus strains in cultured cells. Arch. Virol. 2009, 154, 1945–1951. [Google Scholar] [CrossRef]
- Feng, Z.; Hensley, L.; McKnight, K.L.; Hu, F.; Madden, V.; Ping, L.; Jeong, S.H.; Walker, C.; Lanford, R.E.; Lemon, S.M. A pathogenic picornavirus acquires an envelope by hijacking cellular membranes. Nature 2013, 496, 367–371. [Google Scholar] [CrossRef] [Green Version]
- Geng, Y.; Yang, J.; Huang, W.; Harrison, T.J.; Zhou, Y.; Wen, Z.; Wang, Y. Virus host protein interaction network analysis reveals that the HEV ORF3 protein may interrupt the blood coagulation process. PLoS ONE 2013, 8, e56320. [Google Scholar] [CrossRef] [Green Version]
- Subramani, C.; Nair, V.P.; Anang, S.; Mandal, S.D.; Pareek, M.; Kaushik, N.; Srivastava, A.; Saha, S.; Shalimar; Nayak, B.; et al. Host-virus protein interaction network reveals the involvement of multiple host processes in the life cycle of hepatitis E virus. mSystems 2018, 3, e00135-17. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Li, S.; Yang, C.; Wei, M.; Song, C.; Zheng, Z.; Gu, Y.; Du, H.; Zhang, J.; Xia, N. Homology model and potential virus-capsid binding site of a putative HEV receptor Grp78. J. Mol. Model. 2011, 17, 987–995. [Google Scholar] [CrossRef]
- Ahmed, Z.; Holla, P.; Ahmad, I.; Jameel, S. The ATP synthase subunit β (ATP5B) is an entry factor for the hepatitis E virus. bioRxiv 2016. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Tian, Y.; Wen, Z.; Zhang, F.; Qi, Y.; Huang, W.; Zhang, H.; Wang, Y. Asialoglycoprotein receptor facilitates infection of PLC/PRF/5 cells by HEV through interaction with ORF2. J. Med. Virol. 2016, 88, 2186–2195. [Google Scholar] [CrossRef]
- Kalia, M.; Chandra, V.; Rahman, S.A.; Sehgal, D.; Jameel, S. Heparan sulfate proteoglycans are required for cellular binding of the hepatitis E virus ORF2 capsid protein and for viral infection. J. Virol. 2009, 83, 12714–12724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, X.; Ambardekar, C.; Lu, Y.; Feng, Z. Distinct entry mechanisms for nonenveloped and quasi-enveloped hepatitis E viruses. J. Virol. 2016, 90, 4232–4242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- WuDunn, D.; Spear, P.G. Initial interaction of herpes simplex virus with cells is binding to heparan sulfate. J. Virol. 1989, 63, 52–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shieh, M.T.; WuDunn, D.; Montgomery, R.I.; Esko, J.D.; Spear, P.G. Cell surface receptors for herpes simplex virus are heparan sulfate proteoglycans. J. Cell Biol. 1992, 116, 1273–1281. [Google Scholar] [CrossRef] [PubMed]
- Mondor, I.; Ugolini, S.; Sattentau, Q.J. Human immunodeficiency virus type 1 attachment to HeLa CD4 cells is CD4 independent and gp120 dependent and requires cell surface heparans. J. Virol. 1998, 72, 3623–3634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barth, H.; Schafer, C.; Adah, M.I.; Zhang, F.; Linhardt, R.J.; Toyoda, H.; Kinoshita-Toyoda, A.; Toida, T.; Van Kuppevelt, T.H.; Depla, E.; et al. Cellular binding of hepatitis C virus envelope glycoprotein E2 requires cell surface heparan sulfate. J. Biol. Chem. 2003, 278, 41003–41012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamura, M.; Natori, K.; Kobayashi, M.; Miyamura, T.; Takeda, N. Genogroup II noroviruses efficiently bind to heparan sulfate proteoglycan associated with the cellular membrane. J. Virol. 2004, 78, 3817–3826. [Google Scholar] [CrossRef] [Green Version]
- Shiota, T.; Li, T.C.; Nishimura, Y.; Yoshizaki, S.; Sugiyama, R.; Shimojima, M.; Saijo, M.; Shimizu, H.; Suzuki, R.; Wakita, T.; et al. Integrin alpha3 is involved in non-enveloped hepatitis E virus infection. Virology 2019, 536, 119–124. [Google Scholar] [CrossRef]
- Takada, Y.; Ye, X.; Simon, S. The integrins. Genome Biol. 2007, 8, 215. [Google Scholar] [CrossRef] [Green Version]
- Akula, S.M.; Pramod, N.P.; Wang, F.Z.; Chandran, B. Integrin alpha3beta1 (CD 49c/29) is a cellular receptor for Kaposi’s sarcoma-associated herpesvirus (KSHV/HHV-8) entry into the target cells. Cell 2002, 108, 407–419. [Google Scholar] [CrossRef] [Green Version]
- Rivera-Serrano, E.E.; González-López, O.; Das, A.; Lemon, S.M. Cellular entry and uncoating of naked and quasi-enveloped human hepatoviruses. Elife 2019, 8, e43983. [Google Scholar] [CrossRef] [PubMed]
- Choy, M.Y.; Richman, P.I.; Horton, M.A.; MacDonald, T.T. Expression of the VLA family of integrins in human intestine. J. Pathol. 1990, 160, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Marion, O.; Lhomme, S.; Nayrac, M.; Dubois, M.; Pucelle, M.; Requena, M.; Migueres, M.; Abravanel, F.; Peron, J.M.; Carrere, N.; et al. Hepatitis E virus replication in human intestinal cells. Gut 2020, 69, 901–910. [Google Scholar] [CrossRef] [PubMed]
- Oechslin, N.; Moradpour, D.; Gouttenoire, J. Hepatitis E virus finds its path through the gut. Gut 2020, 69, 796–798. [Google Scholar] [CrossRef]
- de Melker, A.A.; Sterk, L.M.; Delwel, G.O.; Fles, D.L.; Daams, H.; Weening, J.J.; Sonnenberg, A. The A and B variants of the alpha 3 integrin subunit: Tissue distribution and functional characterization. Lab. Invest. 1997, 76, 547–563. [Google Scholar]
- Volpes, R.; van den Oord, J.J.; Desmet, V.J. Distribution of the VLA family of integrins in normal and pathological human liver tissue. Gastroenterology 1991, 101, 200–206. [Google Scholar] [CrossRef]
- Volpes, R.; van den Oord, J.J.; Desmet, V.J. Integrins as differential cell lineage markers of primary liver tumors. Am. J. Pathol. 1993, 142, 1483–1492. [Google Scholar]
- Kapur, N.; Thakral, D.; Durgapal, H.; Panda, S.K. Hepatitis E virus enters liver cells through receptor-dependent clathrin-mediated endocytosis. J. Viral Hepat. 2012, 19, 436–448. [Google Scholar] [CrossRef]
- Holla, P.; Ahmad, I.; Ahmed, Z.; Jameel, S. Hepatitis E virus enters liver cells through a dynamin-2, clathrin and membrane cholesterol-dependent pathway. Traffic 2015, 16, 398–416. [Google Scholar] [CrossRef]
- Bubeck, D.; Filman, D.J.; Cheng, N.; Steven, A.C.; Hogle, J.M.; Belnap, D.M. The structure of the poliovirus 135S cell entry intermediate at 10-angstrom resolution reveals the location of an externalized polypeptide that binds to membranes. J. Virol. 2005, 79, 7745–7755. [Google Scholar] [CrossRef] [Green Version]
- Cavaldesi, M.; Caruso, M.; Sthandier, O.; Amati, P.; Garcia, M.I. Conformational changes of murine polyomavirus capsid proteins induced by sialic acid binding. J. Biol. Chem. 2004, 279, 41573–41579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sapp, M.; Bienkowska-Haba, M. Viral entry mechanisms: human papillomavirus and a long journey from extracellular matrix to the nucleus. FEBS J. 2009, 276, 7206–7216. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Feng, Z. Hepatitis E virus entry. Viruses 2019, 11, 883. [Google Scholar] [CrossRef] [Green Version]
- Tyagi, S.; Korkaya, H.; Zafrullah, M.; Jameel, S.; Lal, S.K. The phosphorylated form of the ORF3 protein of hepatitis E virus interacts with its non-glycosylated form of the major capsid protein, ORF2. J. Biol. Chem. 2002, 277, 22759–22767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drave, S.A.; Debing, Y.; Walter, S.; Todt, D.; Engelmann, M.; Friesland, M.; Wedemeyer, H.; Neyts, J.; Behrendt, P.; Steinmann, E. Extra-hepatic replication and infection of hepatitis E virus in neuronal-derived cells. J. Viral Hepat. 2016, 23, 512–521. [Google Scholar] [CrossRef]
- Fu, R.M.; Decker, C.C.; Dao Thi, V.L. Cell culture models for hepatitis E virus. Viruses 2019, 11, 608. [Google Scholar] [CrossRef] [Green Version]
- Gouilly, J.; Chen, Q.; Siewiera, J.; Cartron, G.; Levy, C.; Dubois, M.; Al-Daccak, R.; Izopet, J.; Jabrane-Ferrat, N.; El Costa, H. Genotype specific pathogenicity of hepatitis E virus at the human maternal-fetal interface. Nat. Commun. 2018, 9, 4748. [Google Scholar] [CrossRef] [Green Version]
- Knegendorf, L.; Drave, S.A.; Dao Thi, V.L.; Debing, Y.; Brown, R.J.P.; Vondran, F.W.R.; Resner, K.; Friesland, M.; Khera, T.; Engelmann, M.; et al. Hepatitis E virus replication and interferon responses in human placental cells. Hepatol. Commun. 2018, 2, 173–187. [Google Scholar] [CrossRef] [Green Version]
- Meister, T.L.; Bruening, J.; Todt, D.; Steinmann, E. Cell culture systems for the study of hepatitis E virus. Antiviral Res. 2019, 163, 34–49. [Google Scholar] [CrossRef]
- Shukla, P.; Nguyen, H.T.; Torian, U.; Engle, R.E.; Faulk, K.; Dalton, H.R.; Bendall, R.P.; Keane, F.E.; Purcell, R.H.; Emerson, S.U. Cross-species infections of cultured cells by hepatitis E virus and discovery of an infectious virus-host recombinant. Proc/ Natl. Acad. Sci. USA 2011, 108, 2438–2443. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Huang, F.; Xu, L.; Lin, Z.; de Vrij, F.M.S.; Ayo-Martin, A.C.; van der Kroeg, M.; Zhao, M.; Yin, Y.; Wang, W.; et al. Hepatitis E virus infects neurons and brains. J. Infect. Dis. 2017, 215, 1197–1206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamar, N.; Bendall, R.; Legrand-Abravanel, F.; Xia, N.S.; Ijaz, S.; Izopet, J.; Dalton, H.R. Hepatitis E. Lancet 2012, 379, 2477–2488. [Google Scholar] [CrossRef]
- Debing, Y.; Moradpour, D.; Neyts, J.; Gouttenoire, J. Update on hepatitis E virology: Implications for clinical practice. J. Hepatol. 2016, 65, 200–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geng, Y.; Zhao, C.; Huang, W.; Harrison, T.J.; Zhang, H.; Geng, K.; Wang, Y. Detection and assessment of infectivity of hepatitis E virus in urine. J. Hepatol. 2016, 64, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Kamar, N.; Abravanel, F.; Lhomme, S.; Rostaing, L.; Izopet, J. Hepatitis E virus: Chronic infection, extra-hepatic manifestations, and treatment. Clin. Res. Hepatol. Gastroenterol. 2015, 39, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Montero, H.; Perez-Gil, G.; Sampieri, C.L. Eukaryotic initiation factor 4A (eIF4A) during viral infections. Virus Genes 2019, 55, 267–273. [Google Scholar] [CrossRef]
- Zhou, X.; Xu, L.; Wang, Y.; Wang, W.; Sprengers, D.; Metselaar, H.J.; Peppelenbosch, M.P.; Pan, Q. Requirement of the eukaryotic translation initiation factor 4F complex in hepatitis E virus replication. Antiviral Res. 2015, 124, 11–19. [Google Scholar] [CrossRef]
- Glitscher, M.; Himmelsbach, K.; Woytinek, K.; Johne, R.; Reuter, A.; Spiric, J.; Schwaben, L.; Grunweller, A.; Hildt, E. Inhibition of hepatitis E virus spread by the natural compound silvestrol. Viruses 2018, 10, 301. [Google Scholar] [CrossRef] [Green Version]
- Todt, D.; Moeller, N.; Praditya, D.; Kinast, V.; Friesland, M.; Engelmann, M.; Verhoye, L.; Sayed, I.M.; Behrendt, P.; Dao Thi, V.L.; et al. The natural compound silvestrol inhibits hepatitis E virus (HEV) replication in vitro and in vivo. Antiviral Res. 2018, 157, 151–158. [Google Scholar] [CrossRef]
- Biedenkopf, N.; Lange-Grunweller, K.; Schulte, F.W.; Weisser, A.; Muller, C.; Becker, D.; Becker, S.; Hartmann, R.K.; Grunweller, A. The natural compound silvestrol is a potent inhibitor of Ebola virus replication. Antiviral Res. 2017, 137, 76–81. [Google Scholar] [CrossRef]
- Elgner, F.; Sabino, C.; Basic, M.; Ploen, D.; Grunweller, A.; Hildt, E. Inhibition of Zika virus replication by silvestrol. Viruses 2018, 10, 149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henss, L.; Scholz, T.; Grunweller, A.; Schnierle, B.S. Silvestrol inhibits Chikungunya virus replication. Viruses 2018, 10, 592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, C.; Schulte, F.W.; Lange-Grunweller, K.; Obermann, W.; Madhugiri, R.; Pleschka, S.; Ziebuhr, J.; Hartmann, R.K.; Grunweller, A. Broad-spectrum antiviral activity of the eIF4A inhibitor silvestrol against corona- and picornaviruses. Antiviral Res. 2018, 150, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Karpe, Y.A.; Lole, K.S. Deubiquitination activity associated with hepatitis E virus putative papain-like cysteine protease. J. Gen. Virol. 2011, 92, 2088–2092. [Google Scholar] [CrossRef]
- Li, C.; Debing, Y.; Jankevicius, G.; Neyts, J.; Ahel, I.; Coutard, B.; Canard, B. Viral macro domains reverse protein ADP-ribosylation. J. Virol. 2016, 90, 8478–8486. [Google Scholar] [CrossRef] [Green Version]
- Kanade, G.D.; Pingale, K.D.; Karpe, Y.A. Activities of thrombin and factor Xa are essential for replication of hepatitis E virus and are possibly implicated in ORF1 polyprotein processing. J. Virol. 2018, 92, e01853-17. [Google Scholar] [CrossRef] [Green Version]
- LeDesma, R.; Nimgaonkar, I.; Ploss, A. Hepatitis E virus replication. Viruses 2019, 11, 719. [Google Scholar] [CrossRef] [Green Version]
- Szkolnicka, D.; Pollan, A.; Da Silva, N.; Oechslin, N.; Gouttenoire, J.; Moradpour, D. Recombinant hepatitis E viruses harboring tags in the ORF1 protein. J. Virol. 2019, 93, e00459-19. [Google Scholar] [CrossRef] [Green Version]
- Ding, Q.; Nimgaonkar, I.; Archer, N.F.; Bram, Y.; Heller, B.; Schwartz, R.E.; Ploss, A. Identification of the intragenomic promoter controlling hepatitis E virus subgenomic RNA transcription. mBio 2018, 9, e00769-18. [Google Scholar] [CrossRef] [Green Version]
- Kanade, G.D.; Pingale, K.D.; Karpe, Y.A. Protein interactions network of hepatitis E virus RNA and polymerase with host proteins. Front. Microbiol. 2019, 10, 2501. [Google Scholar] [CrossRef]
- Pingale, K.D.; Kanade, G.D.; Karpe, Y.A. Heterogeneous nuclear ribonucleoproteins participate in hepatitis E virus (HEV) replication. J. Mol. Biol. 2020, 432, 2369–2387. [Google Scholar] [CrossRef] [PubMed]
- Burnham, A.J.; Gong, L.; Hardy, R.W. Heterogeneous nuclear ribonuclear protein K interacts with Sindbis virus nonstructural proteins and viral subgenomic mRNA. Virology 2007, 367, 212–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettit Kneller, E.L.; Connor, J.H.; Lyles, D.S. hnRNPs relocalize to the cytoplasm following infection with vesicular stomatitis virus. J. Virol. 2009, 83, 770–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bourai, M.; Lucas-Hourani, M.; Gad, H.H.; Drosten, C.; Jacob, Y.; Tafforeau, L.; Cassonnet, P.; Jones, L.M.; Judith, D.; Couderc, T.; et al. Mapping of Chikungunya virus interactions with host proteins identified nsP2 as a highly connected viral component. J. Virol. 2012, 86, 3121–3134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunetti, J.E.; Scolaro, L.A.; Castilla, V. The heterogeneous nuclear ribonucleoprotein K (hnRNP K) is a host factor required for dengue virus and Junin virus multiplication. Virus Res. 2015, 203, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Poenisch, M.; Metz, P.; Blankenburg, H.; Ruggieri, A.; Lee, J.Y.; Rupp, D.; Rebhan, I.; Diederich, K.; Kaderali, L.; Domingues, F.S.; et al. Identification of hnRNPK as regulator of hepatitis C virus particle production. PLoS Pathog. 2015, 11, e1004573. [Google Scholar] [CrossRef]
- Paul, D.; Madan, V.; Bartenschlager, R. Hepatitis C virus RNA replication and assembly: Living on the fat of the land. Cell Host Microbe 2014, 16, 569–579. [Google Scholar] [CrossRef] [Green Version]
- Moradpour, D.; Penin, F.; Rice, C.M. Replication of hepatitis C virus. Nature Rev. Microbiol. 2007, 5, 453–463. [Google Scholar] [CrossRef]
- Farhat, R.; Seron, K.; Ferlin, J.; Feneant, L.; Belouzard, S.; Goueslain, L.; Jackson, C.L.; Dubuisson, J.; Rouillé, Y. Identification of class II ADP-ribosylation factors as cellular factors required for hepatitis C virus replication. Cell. Microbiol. 2016, 18, 1121–1133. [Google Scholar] [CrossRef] [Green Version]
- Goueslain, L.; Alsaleh, K.; Horellou, P.; Roingeard, P.; Descamps, V.; Duverlie, G.; Ciczora, Y.; Wychowski, C.; Dubuisson, J.; Rouille, Y. Identification of GBF1 as a cellular factor required for hepatitis C virus RNA replication. J. Virol. 2010, 84, 773–787. [Google Scholar] [CrossRef] [Green Version]
- Belov, G.A.; Feng, Q.; Nikovics, K.; Jackson, C.L.; Ehrenfeld, E. A critical role of a cellular membrane traffic protein in poliovirus RNA replication. PLoS Pathog. 2008, 4, e1000216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanke, K.H.; van der Schaar, H.M.; Belov, G.A.; Feng, Q.; Duijsings, D.; Jackson, C.L.; Ehrenfeld, E.; van Kuppeveld, F.J. GBF1, a guanine nucleotide exchange factor for Arf, is crucial for coxsackievirus B3 RNA replication. J. Virol. 2009, 83, 11940–11949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Du, J.; Jin, Q. Class I ADP-ribosylation factors are involved in enterovirus 71 replication. PLoS ONE 2014, 9, e99768. [Google Scholar] [CrossRef] [PubMed]
- Farhat, R.; Ankavay, M.; Lebsir, N.; Gouttenoire, J.; Jackson, C.L.; Wychowski, C.; Moradpour, D.; Dubuisson, J.; Rouille, Y.; Cocquerel, L. Identification of GBF1 as a cellular factor required for hepatitis E virus RNA replication. Cell. Microbiol. 2018, 20, e12804. [Google Scholar] [CrossRef] [Green Version]
- Verheije, M.H.; Raaben, M.; Mari, M.; Te Lintelo, E.G.; Reggiori, F.; van Kuppeveld, F.J.; Rottier, P.J.; de Haan, C.A. Mouse hepatitis coronavirus RNA replication depends on GBF1-mediated ARF1 activation. PLoS Pathog. 2008, 4, e1000088. [Google Scholar] [CrossRef] [Green Version]
- Xing, L.; Li, T.C.; Mayazaki, N.; Simon, M.N.; Wall, J.S.; Moore, M.; Wang, C.Y.; Takeda, N.; Wakita, T.; Miyamura, T.; et al. Structure of hepatitis E virion-sized particle reveals an RNA-dependent viral assembly pathway. J. Biol. Chem. 2010, 285, 33175–33183. [Google Scholar] [CrossRef] [Green Version]
- Mori, Y.; Matsuura, Y. Structure of hepatitis E viral particle. Virus Res. 2011, 161, 59–64. [Google Scholar] [CrossRef]
- Graff, J.; Zhou, Y.H.; Torian, U.; Nguyen, H.; St Claire, M.; Yu, C.; Purcell, R.H.; Emerson, S.U. Mutations within potential glycosylation sites in the capsid protein of hepatitis E virus prevent the formation of infectious virus particles. J. Virol. 2008, 82, 1185–1194. [Google Scholar] [CrossRef] [Green Version]
- Montpellier, C.; Wychowski, C.; Sayed, I.M.; Meunier, J.C.; Saliou, J.M.; Ankavay, M.; Bull, A.; Pillez, A.; Abravanel, F.; Helle, F.; et al. Hepatitis E virus lifecycle and identification of 3 forms of the ORF2 capsid protein. Gastroenterology 2018, 154, 211.e218–223.e218. [Google Scholar] [CrossRef]
- Yin, X.; Ying, D.; Lhomme, S.; Tang, Z.; Walker, C.M.; Xia, N.; Zheng, Z.; Feng, Z. Origin, antigenicity, and function of a secreted form of ORF2 in hepatitis E virus infection. Proc. Natl. Acad. Sci. USA 2018, 115, 4773–4778. [Google Scholar] [CrossRef] [Green Version]
- Ankavay, M.; Montpellier, C.; Sayed, I.M.; Saliou, J.M.; Wychowski, C.; Saas, L.; Duvet, S.; Aliouat-Denis, C.M.; Farhat, R.; de Masson d’Autume, V.; et al. New insights into the ORF2 capsid protein, a key player of the hepatitis E virus lifecycle. Sci. Rep. 2019, 9, 6243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenggenhager, D.; Gouttenoire, J.; Malehmir, M.; Bawohl, M.; Honcharova-Biletska, H.; Kreutzer, S.; Semela, D.; Neuweiler, J.; Hurlimann, S.; Aepli, P.; et al. Visualization of hepatitis E virus RNA and proteins in the human liver. J. Hepatol. 2017, 67, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Jameel, S.; Zafrullah, M.; Ozdener, M.H.; Panda, S.K. Expression in animal cells and characterization of the hepatitis E virus structural proteins. J. Virol. 1996, 70, 207–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zafrullah, M.; Ozdener, M.H.; Kumar, R.; Panda, S.K.; Jameel, S. Mutational analysis of glycosylation, membrane translocation, and cell surface expression of the hepatitis E virus ORF2 protein. J. Virol. 1999, 73, 4074–4082. [Google Scholar] [CrossRef] [Green Version]
- Emerson, S.U.; Nguyen, H.; Torian, U.; Purcell, R.H. ORF3 protein of hepatitis E virus is not required for replication, virion assembly, or infection of hepatoma cells in vitro. J. Virol. 2006, 80, 10457–10464. [Google Scholar] [CrossRef] [Green Version]
- Yamada, K.; Takahashi, M.; Hoshino, Y.; Takahashi, H.; Ichiyama, K.; Nagashima, S.; Tanaka, T.; Okamoto, H. ORF3 protein of hepatitis E virus is essential for virion release from infected cells. J. Gen. Virol. 2009, 90, 1880–1891. [Google Scholar] [CrossRef]
- Capelli, N.; Marion, O.; Dubois, M.; Allart, S.; Bertrand-Michel, J.; Lhomme, S.; Abravanel, F.; Izopet, J.; Chapuy-Regaud, S. Vectorial release of hepatitis E virus in polarized human hepatocytes. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [Green Version]
- Dao Thi, V.L.; Wu, X.; Belote, R.L.; Andreo, U.; Takacs, C.N.; Fernandez, J.P.; Vale-Silva, L.A.; Prallet, S.; Decker, C.C.; Fu, R.M.; et al. Stem cell-derived polarized hepatocytes. Nat. Commun. 2020, 11, 1677. [Google Scholar] [CrossRef] [Green Version]
- Zafrullah, M.; Ozdener, M.H.; Panda, S.K.; Jameel, S. The ORF3 protein of hepatitis E virus is a phosphoprotein that associates with the cytoskeleton. J. Virol. 1997, 71, 9045–9053. [Google Scholar] [CrossRef] [Green Version]
- Surjit, M.; Oberoi, R.; Kumar, R.; Lal, S.K. Enhanced alpha1 microglobulin secretion from hepatitis E virus ORF3-expressing human hepatoma cells is mediated by the tumor susceptibility gene 101. J. Biol. Chem. 2006, 281, 8135–8142. [Google Scholar] [CrossRef] [Green Version]
- Nagashima, S.; Takahashi, M.; Jirintai; Tanaka, T.; Yamada, K.; Nishizawa, T.; Okamoto, H. A PSAP motif in the ORF3 protein of hepatitis E virus is necessary for virion release from infected cells. J. Gen. Virol. 2011, 92, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Nagashima, S.; Takahashi, M.; Jirintai, S.; Tanaka, T.; Nishizawa, T.; Yasuda, J.; Okamoto, H. Tumour susceptibility gene 101 and the vacuolar protein sorting pathway are required for the release of hepatitis E virions. J. Gen. Virol. 2011, 92, 2838–2848. [Google Scholar] [CrossRef] [PubMed]
- Garrus, J.E.; von Schwedler, U.K.; Pornillos, O.W.; Morham, S.G.; Zavitz, K.H.; Wang, H.E.; Wettstein, D.A.; Stray, K.M.; Cote, M.; Rich, R.L.; et al. Tsg101 and the vacuolar protein sorting pathway are essential for HIV-1 budding. Cell 2001, 107, 55–65. [Google Scholar] [CrossRef] [Green Version]
- Martin-Serrano, J.; Zang, T.; Bieniasz, P.D. HIV-1 and Ebola virus encode small peptide motifs that recruit Tsg101 to sites of particle assembly to facilitate egress. Nat. Med. 2001, 7, 1313–1319. [Google Scholar] [CrossRef] [PubMed]
- Vietri, M.; Radulovic, M.; Stenmark, H. The many functions of ESCRTs. Nat. Rev. Mol. Cell Biol. 2020, 21, 25–42. [Google Scholar] [CrossRef] [PubMed]
- Nagashima, S.; Jirintai, S.; Takahashi, M.; Kobayashi, T.; Tanggis; Nishizawa, T.; Kouki, T.; Yashiro, T.; Okamoto, H. Hepatitis E virus egress depends on the exosomal pathway, with secretory exosomes derived from multivesicular bodies. J. Gen. Virol. 2014, 95, 2166–2175. [Google Scholar] [CrossRef]
- Nagashima, S.; Takahashi, M.; Jirintai, S.; Tanggis; Kobayashi, T.; Nishizawa, T.; Okamoto, H. The membrane on the surface of hepatitis E virus particles is derived from the intracellular membrane and contains trans-Golgi network protein 2. Arch. Virol. 2014, 159, 979–991. [Google Scholar] [CrossRef]
- Nagashima, S.; Takahashi, M.; Kobayashi, T.; Nishizawa, T.; Nishiyama, T.; Primadharsini, P.P.; Okamoto, H. Characterization of the quasi-enveloped hepatitis E virus particles released by the cellular exosomal pathway. J. Virol. 2017, 91, e00822-17. [Google Scholar] [CrossRef] [Green Version]
- Qi, Y.; Zhang, F.; Zhang, L.; Harrison, T.J.; Huang, W.; Zhao, C.; Kong, W.; Jiang, C.; Wang, Y. Hepatitis E virus produced from cell culture has a lipid envelope. PLoS ONE 2015, 10, e0132503. [Google Scholar] [CrossRef] [Green Version]
- Gouttenoire, J.; Pollan, A.; Abrami, L.; Oechslin, N.; Mauron, J.; Matter, M.; Oppliger, J.; Szkolnicka, D.; Dao Thi, V.L.; van der Goot, F.G.; et al. Palmitoylation mediates membrane association of hepatitis E virus ORF3 protein and is required for infectious particle secretion. PLoS Pathog. 2018, 14, e1007471. [Google Scholar] [CrossRef]
- Blaskovic, S.; Blanc, M.; van der Goot, F.G. What does S-palmitoylation do to membrane proteins? FEBS J. 2013, 280, 2766–2774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapuy-Regaud, S.; Dubois, M.; Plisson-Chastang, C.; Bonnefois, T.; Lhomme, S.; Bertrand-Michel, J.; You, B.; Simoneau, S.; Gleizes, P.E.; Flan, B.; et al. Characterization of the lipid envelope of exosome encapsulated HEV particles protected from the immune response. Biochimie 2017, 141, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Chahar, H.S.; Bao, X.; Casola, A. Exosomes and their role in the life cycle and pathogenesis of RNA viruses. Viruses 2015, 7, 3204–3225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Zhou, X.; Debing, Y.; Chen, K.; Van Der Laan, L.J.; Neyts, J.; Janssen, H.L.; Metselaar, H.J.; Peppelenbosch, M.P.; Pan, Q. Calcineurin inhibitors stimulate and mycophenolic acid inhibits replication of hepatitis E virus. Gastroenterology 2014, 146, 1775–1783. [Google Scholar] [CrossRef]
- Wu, X.; Dao Thi, V.L.; Liu, P.; Takacs, C.N.; Xiang, K.; Andrus, L.; Gouttenoire, J.; Moradpour, D.; Rice, C.M. Pan-genotype hepatitis E virus replication in stem cell-derived hepatocellular systems. Gastroenterology 2018, 154, 663.e7–674.e7. [Google Scholar] [CrossRef]
- Meertens, L.; Hafirassou, M.L.; Couderc, T.; Bonnet-Madin, L.; Kril, V.; Kummerer, B.M.; Labeau, A.; Brugier, A.; Simon-Loriere, E.; Burlaud-Gaillard, J.; et al. FHL1 is a major host factor for Chikungunya virus infection. Nature 2019, 574, 259–263. [Google Scholar] [CrossRef]
- Zhang, R.; Kim, A.S.; Fox, J.M.; Nair, S.; Basore, K.; Klimstra, W.B.; Rimkunas, R.; Fong, R.H.; Lin, H.; Poddar, S.; et al. Mxra8 is a receptor for multiple arthritogenic alphaviruses. Nature 2018, 557, 570–574. [Google Scholar] [CrossRef]
- Han, J.; Perez, J.T.; Chen, C.; Li, Y.; Benitez, A.; Kandasamy, M.; Lee, Y.; Andrade, J.; tenOever, B.; Manicassamy, B. Genome-wide CRISPR/Cas9 screen identifies host factors essential for influenza virus replication. Cell Rep. 2018, 23, 596–607. [Google Scholar] [CrossRef] [Green Version]
- Richardson, R.B.; Ohlson, M.B.; Eitson, J.L.; Kumar, A.; McDougal, M.B.; Boys, I.N.; Mar, K.B.; De La Cruz-Rivera, P.C.; Douglas, C.; Konopka, G.; et al. A CRISPR screen identifies IFI6 as an ER-resident interferon effector that blocks flavivirus replication. Nat. Microbiol. 2018, 3, 1214–1223. [Google Scholar] [CrossRef] [Green Version]
- Flint, M.; Chatterjee, P.; Lin, D.L.; McMullan, L.K.; Shrivastava-Ranjan, P.; Bergeron, E.; Lo, M.K.; Welch, S.R.; Nichol, S.T.; Tai, A.W.; et al. A genome-wide CRISPR screen identifies N-acetylglucosamine-1-phosphate transferase as a potential antiviral target for Ebola virus. Nat. Commun. 2019, 10, 285. [Google Scholar] [CrossRef] [Green Version]
- V’Kovski, P.; Gerber, M.; Kelly, J.; Pfaender, S.; Ebert, N.; Braga Lagache, S.; Simillion, C.; Portmann, J.; Stalder, H.; Gaschen, V.; et al. Determination of host proteins composing the microenvironment of coronavirus replicase complexes by proximity-labeling. Elife 2019, 8, e42037. [Google Scholar] [CrossRef] [PubMed]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oechslin, N.; Moradpour, D.; Gouttenoire, J. On the Host Side of the Hepatitis E Virus Life Cycle. Cells 2020, 9, 1294. https://doi.org/10.3390/cells9051294
Oechslin N, Moradpour D, Gouttenoire J. On the Host Side of the Hepatitis E Virus Life Cycle. Cells. 2020; 9(5):1294. https://doi.org/10.3390/cells9051294
Chicago/Turabian StyleOechslin, Noémie, Darius Moradpour, and Jérôme Gouttenoire. 2020. "On the Host Side of the Hepatitis E Virus Life Cycle" Cells 9, no. 5: 1294. https://doi.org/10.3390/cells9051294
APA StyleOechslin, N., Moradpour, D., & Gouttenoire, J. (2020). On the Host Side of the Hepatitis E Virus Life Cycle. Cells, 9(5), 1294. https://doi.org/10.3390/cells9051294