The Pluripotency Factor Nanog Protects against Neuronal Amyloid β-Induced Toxicity and Oxidative Stress through Insulin Sensitivity Restoration

 , ,
, , 
Abstract
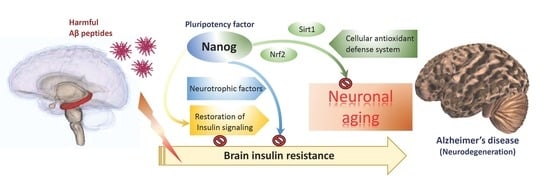
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture, Transfection and Viability Assay
2.3. mRNA Expression Analysis by Reverse-Transcription Quantitative PCR (qPCR)
2.4. Western Blot Analysis
2.5. DAPI Staining on Nuclei
2.6. Detection of Superoxide by Dihydroethidium (DHE) Staining
2.7. Analysis of Mitochondrial Membrane Potential by JC-1 Staining
2.8. SA-β-Galactosidase Staining
2.9. Experimental Animals
2.10. T-Maze Test and Object Recognition Behavior Tests
2.11. Statistical Analysis
3. Results
3.1. Overexpression of Nanog Significantly Reduced Aβ-Mediated Cytotoxicity
3.2. The Protection from Aβ-Induced Apoptosis by Nanog is Dependent on Insulin Signaling in SK-N-MC Neuronal Cells
3.3. Aβ-Induced Superoxide Accumulation and Mitochondrial Dysfunction are Attenuated by Nanog Overexpression
3.4. Overexpression of Nanog Protects Rat Brain against Aβ-Induced Cognitive Impairments
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chen, X.Q.; Mobley, W.C. Alzheimer Disease Pathogenesis: Insights From Molecular and Cellular Biology Studies of Oligomeric Abeta and Tau Species. Front. Neurosci. 2019, 13, 659. [Google Scholar] [CrossRef]
- Lin, C.L.; Huang, W.N.; Li, H.H.; Huang, C.N.; Hsieh, S.; Lai, C.; Lu, F.J. Hydrogen-rich water attenuates amyloid beta-induced cytotoxicity through upregulation of Sirt1-FoxO3a by stimulation of AMP-activated protein kinase in SK-N-MC cells. Chem. -Biol. Interact. 2015, 240, 12–21. [Google Scholar] [CrossRef]
- Marchant, N.L.; Reed, B.R.; Sanossian, N.; Madison, C.M.; Kriger, S.; Dhada, R.; Mack, W.J.; DeCarli, C.; Weiner, M.W.; Mungas, D.M.; et al. The aging brain and cognition: Contribution of vascular injury and abeta to mild cognitive dysfunction. Jama Neurol. 2013, 70, 488–495. [Google Scholar] [CrossRef] [Green Version]
- Wei, Z.; Chen, X.C.; Song, Y.; Pan, X.D.; Dai, X.M.; Zhang, J.; Cui, X.L.; Wu, X.L.; Zhu, Y.G. Amyloid beta Protein Aggravates Neuronal Senescence and Cognitive Deficits in 5XFAD Mouse Model of Alzheimer’s Disease. Chin. Med. J. 2016, 129, 1835–1844. [Google Scholar] [CrossRef]
- Mecocci, P.; Boccardi, V.; Cecchetti, R.; Bastiani, P.; Scamosci, M.; Ruggiero, C.; Baroni, M. A Long Journey into Aging, Brain Aging, and Alzheimer’s Disease Following the Oxidative Stress Tracks. J. Alzheimer’s Dis. 2018, 62, 1319–1335. [Google Scholar] [CrossRef] [Green Version]
- Sala Frigerio, C.; Wolfs, L.; Fattorelli, N.; Thrupp, N.; Voytyuk, I.; Schmidt, I.; Mancuso, R.; Chen, W.T.; Woodbury, M.E.; Srivastava, G.; et al. The Major Risk Factors for Alzheimer’s Disease: Age, Sex, and Genes Modulate the Microglia Response to Abeta Plaques. Cell Rep. 2019, 27, 1293–1306. [Google Scholar] [CrossRef] [Green Version]
- Gouras, G.K. Aging, Metabolism, Synaptic Activity, and Abeta in Alzheimer’s Disease. Front. Aging Neurosci. 2019, 11, 185. [Google Scholar] [CrossRef] [Green Version]
- Maciel-Baron, L.A.; Moreno-Blas, D.; Morales-Rosales, S.L.; Gonzalez-Puertos, V.Y.; Lopez-Diazguerrero, N.E.; Torres, C.; Castro-Obregon, S.; Konigsberg, M. Cellular Senescence, Neurological Function, and Redox State. Antioxid. Redox Signal. 2018, 28, 1704–1723. [Google Scholar] [CrossRef]
- Fouad, G.I. Stem cells as a promising therapeutic approach for Alzheimer’s disease: A review. Bull. Natl. Res. Cent. 2019, 43, 52. [Google Scholar] [CrossRef]
- Ahmed Nel, M.; Murakami, M.; Hirose, Y.; Nakashima, M. Therapeutic Potential of Dental Pulp Stem Cell Secretome for Alzheimer’s Disease Treatment: An In Vitro Study. Stem. Cells Int. 2016, 2016, 8102478. [Google Scholar] [CrossRef]
- Griffith, C.M.; Eid, T.; Rose, G.M.; Patrylo, P.R. Evidence for altered insulin receptor signaling in Alzheimer’s disease. Neuropharmacology 2018, 136, 202–215. [Google Scholar] [CrossRef]
- Maciejczyk, M.; Zebrowska, E.; Chabowski, A. Insulin Resistance and Oxidative Stress in the Brain: What’s New? Int. J. Mol. Sci. 2019, 20, 874. [Google Scholar] [CrossRef] [Green Version]
- Bedse, G.; Di Domenico, F.; Serviddio, G.; Cassano, T. Aberrant insulin signaling in Alzheimer’s disease: Current knowledge. Front. Neurosci. 2015, 9, 204. [Google Scholar] [CrossRef] [Green Version]
- Li, H.H.; Lin, S.L.; Huang, C.N.; Lu, F.J.; Chiu, P.Y.; Huang, W.N.; Lai, T.J.; Lin, C.L. miR-302 Attenuates Amyloid-beta-Induced Neurotoxicity through Activation of Akt Signaling. J. Alzheimer’s Dis. 2016, 50, 1083–1098. [Google Scholar] [CrossRef]
- Kuijk, E.W.; van Mil, A.; Brinkhof, B.; Penning, L.C.; Colenbrander, B.; Roelen, B.A. PTEN and TRP53 independently suppress Nanog expression in spermatogonial stem cells. Stem. Cells Dev. 2010, 19, 979–988. [Google Scholar] [CrossRef]
- Han, J.; Mistriotis, P.; Lei, P.; Wang, D.; Liu, S.; Andreadis, S.T. Nanog reverses the effects of organismal aging on mesenchymal stem cell proliferation and myogenic differentiation potential. Stem Cells 2012, 30, 2746–2759. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.W.; Chang, C.C.; Chang, T.S.; Li, H.H.; Hung, H.C.; Liu, G.Y.; Lin, C.L. Abeta stimulates microglial activation through antizyme-dependent downregulation of ornithine decarboxylase. J. Cell. Physiol. 2019, 234, 9733–9745. [Google Scholar] [CrossRef]
- Lin, C.L.; Cheng, Y.S.; Li, H.H.; Chiu, P.Y.; Chang, Y.T.; Ho, Y.J.; Lai, T.J. Amyloid-beta suppresses AMP-activated protein kinase (AMPK) signaling and contributes to alpha-synuclein-induced cytotoxicity. Exp. Neurol. 2016, 275 Pt 1, 84–98. [Google Scholar] [CrossRef]
- Huang, C.K.; Chang, Y.T.; Amstislavskaya, T.G.; Tikhonova, M.A.; Lin, C.L.; Hung, C.S.; Lai, T.J.; Ho, Y.J. Synergistic effects of ceftriaxone and erythropoietin on neuronal and behavioral deficits in an MPTP-induced animal model of Parkinson’s disease dementia. Behav. Brain. Res. 2015, 294, 198–207. [Google Scholar] [CrossRef]
- Toral-Rios, D.; Pichardo-Rojas, P.S.; Alonso-Vanegas, M.; Campos-Pena, V. GSK3beta and Tau Protein in Alzheimer’s Disease and Epilepsy. Front. Cell. Neurosci. 2020, 14, 19. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Kostov, R.V.; Kazantsev, A.G. The role of Nrf2 signaling in counteracting neurodegenerative diseases. FEBS J. 2018, 285, 3576–3590. [Google Scholar] [CrossRef] [Green Version]
- Chakravorty, A.; Jetto, C.T.; Manjithaya, R. Dysfunctional Mitochondria and Mitophagy as Drivers of Alzheimer’s Disease Pathogenesis. Front. Aging Neurosci. 2019, 11, 311. [Google Scholar] [CrossRef]
- Martinez-Cue, C.; Rueda, N. Cellular Senescence in Neurodegenerative Diseases. Front. Cell. Neurosci. 2020, 14, 16. [Google Scholar] [CrossRef]
- Miranda, M.; Morici, J.F.; Zanoni, M.B.; Bekinschtein, P. Brain-Derived Neurotrophic Factor: A Key Molecule for Memory in the Healthy and the Pathological Brain. Front. Cell. Neurosci. 2019, 13, 363. [Google Scholar] [CrossRef]
- Pardo, J.; Uriarte, M.; Console, G.M.; Reggiani, P.C.; Outeiro, T.F.; Morel, G.R.; Goya, R.G. Insulin-like growth factor-I gene therapy increases hippocampal neurogenesis, astrocyte branching and improves spatial memory in female aging rats. Eur. J. Neurosci. 2016, 44, 2120–2128. [Google Scholar] [CrossRef]
- Lin, C.L.; Huang, C.N. The neuroprotective effects of the anti-diabetic drug linagliptin against Abeta-induced neurotoxicity. Neural Regen. Res. 2016, 11, 236–237. [Google Scholar] [CrossRef]
- Lee, S.H.; Zabolotny, J.M.; Huang, H.; Lee, H.; Kim, Y.B. Insulin in the nervous system and the mind: Functions in metabolism, memory, and mood. Mol. Metab. 2016, 5, 589–601. [Google Scholar] [CrossRef]
- Duarte, A.I.; Moreira, P.I.; Oliveira, C.R. Insulin in central nervous system: More than just a peripheral hormone. J. Aging Res. 2012, 2012, 384017. [Google Scholar] [CrossRef] [Green Version]
- Spinelli, M.; Fusco, S.; Grassi, C. Brain Insulin Resistance and Hippocampal Plasticity: Mechanisms and Biomarkers of Cognitive Decline. Front. Neurosci. 2019, 13, 788. [Google Scholar] [CrossRef] [Green Version]
- Rad, S.K.; Arya, A.; Karimian, H.; Madhavan, P.; Rizwan, F.; Koshy, S.; Prabhu, G. Mechanism involved in insulin resistance via accumulation of beta-amyloid and neurofibrillary tangles: Link between type 2 diabetes and Alzheimer’s disease. Drug Des. Dev. 2018, 12, 3999–4021. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.C.; Lien, C.C.; Hou, W.H.; Chiang, P.M.; Tsai, K.J. Gain of BDNF Function in Engrafted Neural Stem Cells Promotes the Therapeutic Potential for Alzheimer’s Disease. Sci. Rep. 2016, 6, 27358. [Google Scholar] [CrossRef]
- Hosseini, S.A.; Mohammadi, R.; Noruzi, S.; Mohamadi, Y.; Azizian, M.; Mousavy, S.M.; Ghasemi, F.; Hesari, A.; Sahebkar, A.; Salarinia, R.; et al. Stem cell- and gene-based therapies as potential candidates in Alzheimer’s therapy. J. Cell Biochem. 2018, 119, 8723–8736. [Google Scholar] [CrossRef]
- Apple, D.M.; Solano-Fonseca, R.; Kokovay, E. Neurogenesis in the aging brain. Biochem. Pharm. 2017, 141, 77–85. [Google Scholar] [CrossRef]
- Teng, C.F.; Jeng, L.B.; Shyu, W.C. Role of Insulin-like Growth Factor 1 Receptor Signaling in Stem Cell Stemness and Therapeutic Efficacy. Cell Transpl. 2018, 27, 1313–1319. [Google Scholar] [CrossRef] [Green Version]
- Sarubbo, F.; Moranta, D.; Pani, G. Dietary polyphenols and neurogenesis: Molecular interactions and implication for brain ageing and cognition. Neurosci. Biobehav. Rev. 2018, 90, 456–470. [Google Scholar] [CrossRef]
- Ray, S.; Corenblum, M.J.; Anandhan, A.; Reed, A.; Ortiz, F.O.; Zhang, D.D.; Barnes, C.A.; Madhavan, L. A Role for Nrf2 Expression in Defining the Aging of Hippocampal Neural Stem Cells. Cell Transpl. 2018, 27, 589–606. [Google Scholar] [CrossRef]
- Murphy, K.E.; Park, J.J. Can Co-Activation of Nrf2 and Neurotrophic Signaling Pathway Slow Alzheimer’s Disease? Int. J. Mol. Sci. 2017, 18, 1168. [Google Scholar] [CrossRef] [Green Version]
- Brookhouser, N.; Tekel, S.J.; Standage-Beier, K.; Nguyen, T.; Schwarz, G.; Wang, X.; Brafman, D.A. BIG-TREE: Base-Edited Isogenic hPSC Line Generation Using a Transient Reporter for Editing Enrichment. Stem Cell Rep. 2020, 14, 184–191. [Google Scholar] [CrossRef] [Green Version]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, C.-C.; Li, H.-H.; Tsou, S.-H.; Hung, H.-C.; Liu, G.-Y.; Korolenko, T.A.; Lai, T.-J.; Ho, Y.-J.; Lin, C.-L. The Pluripotency Factor Nanog Protects against Neuronal Amyloid β-Induced Toxicity and Oxidative Stress through Insulin Sensitivity Restoration. Cells 2020, 9, 1339. https://doi.org/10.3390/cells9061339
Chang C-C, Li H-H, Tsou S-H, Hung H-C, Liu G-Y, Korolenko TA, Lai T-J, Ho Y-J, Lin C-L. The Pluripotency Factor Nanog Protects against Neuronal Amyloid β-Induced Toxicity and Oxidative Stress through Insulin Sensitivity Restoration. Cells. 2020; 9(6):1339. https://doi.org/10.3390/cells9061339
Chicago/Turabian StyleChang, Ching-Chi, Hsin-Hua Li, Sing-Hua Tsou, Hui-Chih Hung, Guang-Yaw Liu, Tatiana A. Korolenko, Te-Jen Lai, Ying-Jui Ho, and Chih-Li Lin. 2020. "The Pluripotency Factor Nanog Protects against Neuronal Amyloid β-Induced Toxicity and Oxidative Stress through Insulin Sensitivity Restoration" Cells 9, no. 6: 1339. https://doi.org/10.3390/cells9061339
APA StyleChang, C. -C., Li, H. -H., Tsou, S. -H., Hung, H. -C., Liu, G. -Y., Korolenko, T. A., Lai, T. -J., Ho, Y. -J., & Lin, C. -L. (2020). The Pluripotency Factor Nanog Protects against Neuronal Amyloid β-Induced Toxicity and Oxidative Stress through Insulin Sensitivity Restoration. Cells, 9(6), 1339. https://doi.org/10.3390/cells9061339