Administration of Human Non-Diabetic Mesenchymal Stromal Cells to a Murine Model of Diabetic Fracture Repair: A Pilot Study

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mesenchymal Stromal Cell Isolation and Culture Expansion
2.2. Mesenchymal Stromal Cell Phenotypic Characterization
2.3. Mesenchymal Stromal Cell Differentiation
2.4. Pre-Clinical Model of Diabetic Fracture Repair
2.5. In Vivo Assessment of Cellular Biodistribution
2.6. Micro-Computed Tomography
2.7. Mechanical Testing
2.8. Xenogeneic Re-stimulation Assays
2.9. Statistical Analysis
3. Results
3.1. Bone Marrow-Derived MSC Characterization
3.2. Murine Model of Diabetic Fracture Repair
3.3. Cellular Retention and Distribution
3.4. MSC Administration did not Augment the Quality of De Novo Reparative Bone
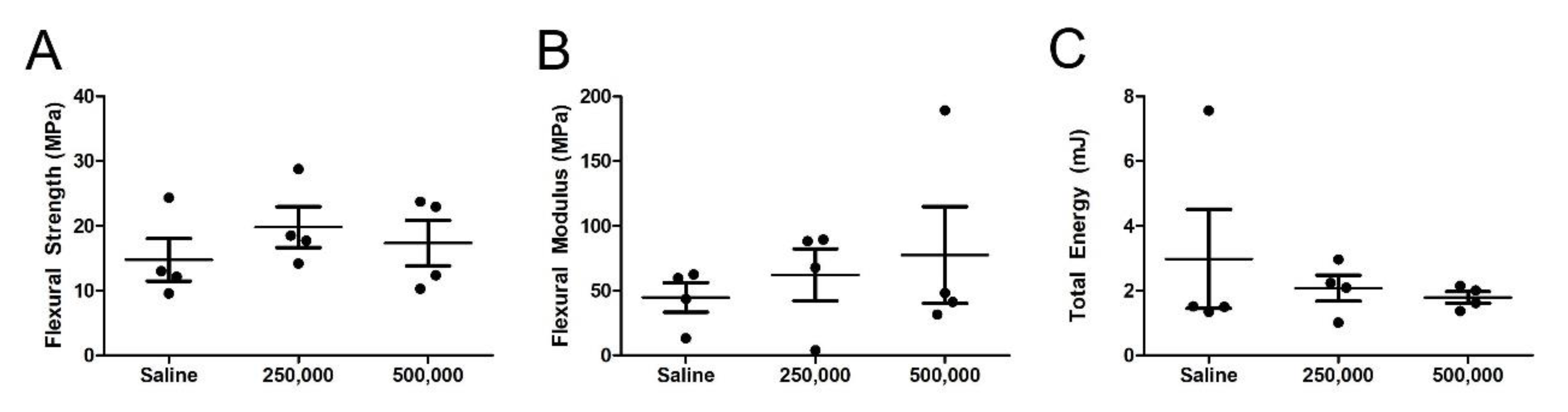
3.5. Mechanical Assessment of the MSC-Treated Fracture
3.6. Xenogeneic MSCs do not Stimulate Lymphocytic Proliferation
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kemink, S.A.G.; Hermus, A.R.M.M.; Swinkels, L.M.J.W.; Lutterman, J.A.; Smals, A.G.H. Osteopenia in insulin-dependent diabetes mellitus; prevalence and aspects of pathophysiology. J. Endocrinol. Investig. 2000, 23, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Thrailkill, K.M. Insulin-Like Growth Factor-I in Diabetes Mellitus: Its Physiology, Metabolic Effects, and Potential Clinical Utility. Diabetes Technol. Ther. 2000, 2, 69–80. [Google Scholar] [CrossRef]
- Janghorbani, M.; Feskanich, D.; Willett, W.C.; Hu, F. Prospective Study of Diabetes and Risk of Hip Fracture: The Nurses’ Health Study. Diabetes Care 2006, 29, 1573–1578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicodemus, K.K.; Folsom, A.R. Type 1 and type 2 diabetes and incident hip fractures in postmenopausal women. Diabetes Care 2001, 24, 1192–1197. [Google Scholar] [CrossRef] [Green Version]
- Loder, R.T. The Influence of Diabetes Mellitus on the Healing of Closed Fractures. Clin. Orthop. Relat. Res. 1988, 210–216. [Google Scholar] [CrossRef]
- Herrero, S.; Calvo, O.; García-Moreno, C.; Martín, E.; Román, J.I.S.; Martín, M.; García-Talavera, J.R.; Calvo, J.J.; Del Pino-Montes, J. Low bone density with normal bone turnover in ovariectomized and streptozotocin-induced diabetic rats. Calcif. Tissue Int. 1998, 62, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Verhaeghe, J.; van Herck, E.; Visser, W.J.; Suiker, A.M.; Thomasset, M.; Einhorn, T.A.; Faierman, E.; Bouillon, R. Bone and mineral metabolism in bb rats with long-term diabetes. Decreased bone turnover and osteoporosis. Diabetes 1990, 39, 477–482. [Google Scholar] [PubMed]
- Silva, M.J.; Brodt, M.D.; Lynch, M.A.; McKenzie, J.A.; Tanouye, K.M.; Nyman, J.S.; Wang, X. Type 1 Diabetes in Young Rats Leads to Progressive Trabecular Bone Loss, Cessation of Cortical Bone Growth, and Diminished Whole Bone Strength and Fatigue Life. J. Bone Miner. Res. 2009, 24, 1618–1627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macey, L.R.; Kana, S.M.; Jingushi, S.; Terek, R.; Borretos, J.; E Bolander, M. Defects of early fracture-healing in experimental diabetes. J. Bone Jt. Surgery-American Vol. 1989, 71, 722–733. [Google Scholar] [CrossRef]
- Ogasawara, A.; Nakajima, A.; Nakajima, F.; Goto, K.-I.; Yamazaki, M. Molecular basis for affected cartilage formation and bone union in fracture healing of the streptozotocin-induced diabetic rat. Bone 2008, 43, 832–839. [Google Scholar] [CrossRef]
- Kayal, R.; Tsatsas, D.; A Bauer, M.; Allen, B.; O Al-Sebaei, M.; Kakar, S.; Leone, C.W.; Morgan, E.F.; Gerstenfeld, L.C.; Einhorn, T.; et al. Diminished bone formation during diabetic fracture healing is related to the premature resorption of cartilage associated with increased osteoclast activity. J. Bone Miner. Res. 2007, 22, 560–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, J.C.; Sampaio, P.; Fernandes, M.H.; Gomes, P.S. The Osteogenic Priming of Mesenchymal Stem Cells is Impaired in Experimental Diabetes. J. Cell. Biochem. 2015, 116, 1658–1667. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, M.; Abu-Shahba, N.; Azmy, O.; El-Badri, N. Impact of Diabetes Mellitus on Human Mesenchymal Stromal Cell Biology and Functionality: Implications for Autologous Transplantation. Stem Cell Rev. Rep. 2019, 15, 194–217. [Google Scholar] [CrossRef] [PubMed]
- Davies, L.; Alm, J.J.; Heldring, N.; Moll, G.; Gavin, C.; Batsis, I.; Qian, H.; Sigvardsson, M.; Nilsson, B.; Kyllonen, L.E.; et al. Type 1 Diabetes Mellitus Donor Mesenchymal Stromal Cells Exhibit Comparable Potency to Healthy Controls In Vitro. STEM CELLS Transl. Med. 2016, 5, 1485–1495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motyl, K.; McCabe, L.R. Streptozotocin, Type I Diabetes Severity and Bone. Boil. Proced. Online 2009, 11, 296–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marturano, J.E.; Cleveland, B.C.; Byrne, M.A.; O’Connell, S.L.; Wixted, J.J.; Billiar, K.L. An improved murine femur fracture device for bone healing studies. J. Biomech. 2008, 41, 1222–1228. [Google Scholar] [CrossRef] [PubMed]
- Creane, M.; Howard, L.; O’Brien, T.E.; Coleman, A.C.M. Biodistribution and retention of locally administered human mesenchymal stromal cells: Quantitative polymerase chain reaction–based detection of human DNA in murine organs. Cytotherapy 2017, 19, 384–394. [Google Scholar] [CrossRef] [Green Version]
- Coleman, A.C.M.; Scheremeta, B.H.; Boyce, A.T.; Mauck, R.L.; Tuan, R.S. Delayed Fracture Healing in Growth Differentiation Factor 5-deficient Mice: A Pilot Study. Clin. Orthop. Relat. Res. 2011, 469, 2915–2924. [Google Scholar] [CrossRef] [Green Version]
- Saffar, K.P.; JamilPour, N.; Rajaai, S.M. How does the bone shaft geometry affect its bending properties? Am. J. Appl. Sci. 2009, 6, 463–470. [Google Scholar] [CrossRef]
- Ryan, A.; Lohan, P.; O’Flynn, L.; Treacy, O.; Chen, X.; Coleman, A.C.M.; Shaw, G.; Murphy, M.; Barry, F.; Griffin, M.D.; et al. Chondrogenic Differentiation Increases Antidonor Immune Response to Allogeneic Mesenchymal Stem Cell Transplantation. Mol. Ther. 2013, 22, 655–667. [Google Scholar] [CrossRef] [Green Version]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.; Krause, D.; Deans, R.; Keating, A.; Prockop, D.; Horwitz, E. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef] [PubMed]
- Botolin, S.; Faugere, M.-C.; Malluche, H.; Orth, M.; Meyer, R.A.; McCabe, L.R. Increased bone adiposity and peroxisomal proliferator-activated receptor-gamma2 expression in type I diabetic mice. Endocrinology 2005, 146, 3622–3631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stolzing, A.; Sellers, D.; Llewelyn, O.; Scutt, A. Diabetes Induced Changes in Rat Mesenchymal Stem Cells. Cells Tissues Organs 2010, 191, 453–465. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-M.; Schilling, T.; Benisch, P.; Zeck, S.; Meissner-Weigl, J.; Schneider, D.; Limbert, C.; Seufert, J.; Kassem, M.; Schütze, N.; et al. Effects of high glucose on mesenchymal stem cell proliferation and differentiation. Biochem. Biophys. Res. Commun. 2007, 363, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Hwang, I.; Hwang, S.H.; Han, H.; Ha, H. Human umbilical cord blood-derived mesenchymal stem cells prevent diabetic renal injury through paracrine action. Diabetes Res. Clin. Pr. 2012, 98, 465–473. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Park, J.; Hwang, S.; Han, H.J.; Ha, H. Delayed Treatment With Human Umbilical Cord Blood-Derived Stem Cells Attenuates Diabetic Renal Injury. Transplant. Proc. 2012, 44, 1123–1126. [Google Scholar] [CrossRef]
- Falanga, V.; Iwamoto, S.; Chartier, M.; Yufit, T.; Butmarc, J.; Kouttab, N.; Shrayer, D.; Carson, P. Autologous Bone Marrow–Derived Cultured Mesenchymal Stem Cells Delivered in a Fibrin Spray Accelerate Healing in Murine and Human Cutaneous Wounds. Tissue Eng. 2007, 13, 1299–1312. [Google Scholar] [CrossRef]
- Scalinci, S.Z.; Scorolli, L.; Corradetti, G.; Domanico, D.; Vingolo, E.M.; Meduri, A.; Bifani, M.; Siravo, D. Potential role of intravitreal human placental stem cell implants in inhibiting progression of diabetic retinopathy in type 2 diabetes: Neuroprotective growth factors in the vitreous. Clin. Ophthalmol. 2011, 5, 691–696. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Li, K.; Yan, X.; Dong, F.; Zhao, R.C. Amelioration of diabetic retinopathy by engrafted human adipose-derived mesenchymal stem cells in streptozotocin diabetic rats. Graefe’s Arch. Clin. Exp. Ophthalmol. 2010, 248, 1415–1422. [Google Scholar] [CrossRef]
- Breitbart, E.A.; Meade, S.; Azad, V.; Yeh, S.; Al-Zube, L.; Lee, Y.-S.; Benevenia, J.; Arinzeh, T.L.; Lin, S.S. Mesenchymal stem cells accelerate bone allograft incorporation in the presence of diabetes mellitus. J. Orthop. Res. 2010, 28, 942–949. [Google Scholar] [CrossRef]
- Niemeyer, P.; Szalay, K.; Luginbühl, R.; Südkamp, N.; Kasten, P. Transplantation of human mesenchymal stem cells in a non-autogenous setting for bone regeneration in a rabbit critical-size defect model. Acta Biomater. 2010, 6, 900–908. [Google Scholar] [CrossRef] [PubMed]
- Szkudelski, T. The mechanism of alloxan and streptozotocin action in B cells of the rat pancreas. Physiol. Res. 2001, 50, 537–546. [Google Scholar] [PubMed]
- Sun, Y.; Chen, L.; Hou, X.; Hou, W.-K.; Dong, J.-J.; Sun, L.; Tang, K.-X.; Wang, B.; Song, J.; Li, H.; et al. Differentiation of bone marrow-derived mesenchymal stem cells from diabetic patients into insulin-producing cells in vitro. Chin. Med. J. 2007, 120, 771–776. [Google Scholar] [CrossRef]
- Dong, Q.-Y.; Chen, L.; Gao, G.-Q.; Wang, L.; Song, J.; Chen, B.; Xu, Y.-X.; Sun, L. Allogeneic diabetic mesenchymal stem cells transplantation in streptozotocin-induced diabetic rat. Clin. Investig. Med. 2008, 31, 328–E337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Q.-P.; Huang, H.; Xu, B.; Dong, X.; Gao, S.-L.; Zhang, B.; Wu, Y.-L. Human bone marrow mesenchymal stem cells differentiate into insulin-producing cells upon microenvironmental manipulation in vitro. Differentiation 2009, 77, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Eggenhofer, E.; Benseler, V.; Kroemer, A.; Popp, F.C.; Geissler, E.K.; Schlitt, H.J.; Baan, C.; Dahlke, M.H.; Hoogduijn, M.J. Mesenchymal stem cells are short-lived and do not migrate beyond the lungs after intravenous infusion. Front. Immunol. 2012, 3, 297. [Google Scholar] [CrossRef] [Green Version]
- Galleu, A.; Riffo-Vasquez, Y.; Trento, C.; Lomas, C.; Dolcetti, L.; Cheung, T.S.; Von Bonin, M.; Barbieri, L.; Halai, K.; Ward, S.; et al. Apoptosis in mesenchymal stromal cells induces in vivo recipient-mediated immunomodulation. Sci. Transl. Med. 2017, 9, eaam7828. [Google Scholar] [CrossRef] [Green Version]
- Khabbal, J.; Kerkelä, E.; Mitkari, B.; Raki, M.; Nystedt, J.; Mikkonen, V.; Bergström, K.; Laitinen, S.; Korhonen, M.; Jolkkonen, J. Differential Clearance of Rat and Human Bone Marrow-Derived Mesenchymal Stem Cells from the Brain after Intra-arterial Infusion in Rats. Cell Transplant. 2015, 24, 819–828. [Google Scholar] [CrossRef] [Green Version]
- Von Bahr, L.; Batsis, I.; Moll, G.; Hagg, M.; Szakos, A.; Sundberg, B.; Uzunel, M.; Ringden, O.; Le Blanc, K. Analysis of Tissues Following Mesenchymal Stromal Cell Therapy in Humans Indicates Limited Long-Term Engraftment and No Ectopic Tissue Formation. STEM CELLS 2012, 30, 1575–1578. [Google Scholar] [CrossRef]
- Pereira, R.F.; Halford, K.W.; O’Hara, M.D.; Leeper, D.B.; Sokolov, B.P.; Pollard, M.D.; Bagasra, O.; Prockop, D.J. Cultured adherent cells from marrow can serve as long-lasting precursor cells for bone, cartilage, and lung in irradiated mice. Proc. Natl. Acad. Sci. 1995, 92, 4857–4861. [Google Scholar] [CrossRef] [Green Version]
- Granero-Moltó, F.; Weis, J.; Miga, M.I.; Landis, B.; Myers, T.J.; O’Rear, L.; Longobardi, L.; Jansen, E.D.; Mortlock, U.P.; Spagnoli, A. Regenerative Effects of Transplanted Mesenchymal Stem Cells in Fracture Healing. STEM CELLS 2009, 27, 1887–1898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nilsson, S.K.; Dooner, M.S.; Weier, H.-U.; Frenkel, B.; Lian, J.B.; Stein, G.S.; Quesenberry, P.J. Cells Capable of Bone Production Engraft from Whole Bone Marrow Transplants in Nonablated Mice. J. Exp. Med. 1999, 189, 729–734. [Google Scholar] [CrossRef] [PubMed]
- Ramot, Y.; Meiron, M.; Toren, A.; Steiner, M.; Nyska, A. Safety and Biodistribution Profile of Placental-derived Mesenchymal Stromal Cells (PLX-PAD) Following Intramuscular Delivery. Toxicol. Pathol. 2009, 37, 606–616. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Xu, L.; Sun, Y.; Zhang, Y.; Li, G. The fate of systemically administrated allogeneic mesenchymal stem cells in mouse femoral fracture healing. Stem Cell Res. Ther. 2015, 6, 206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, C.; Coleman, A.C.M. Impact of Diabetes Mellitus on Bone Health. Int. J. Mol. Sci. 2019, 20, 4873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burdette, A.J.; Guda, T.; Thompson, M.E.; Banas, R.; Sheppard, F. A Novel Secretome Biotherapeutic Influences Regeneration in Critical Size Bone Defects. J. Craniofacial Surg. 2018, 29, 116–123. [Google Scholar] [CrossRef]
- Wang, C.-Y.; Yang, H.-B.; Hsu, H.-S.; Chen, L.-L.; Tsai, C.-C.; Tsai, K.-S.; Yew, T.-L.; Kao, Y.-H.; Hung, S.-C. Mesenchymal stem cell-conditioned medium facilitates angiogenesis and fracture healing in diabetic rats. J. Tissue Eng. Regen. Med. 2011, 6, 559–569. [Google Scholar] [CrossRef]
- Furuta, T.; Miyaki, S.; Ishitobi, H.; Ogura, T.; Kato, Y.; Kamei, N.; Miyado, K.; Higashi, Y.; Ochi, M. Mesenchymal Stem Cell-Derived Exosomes Promote Fracture Healing in a Mouse Model. STEM CELLS Transl. Med. 2016, 5, 1620–1630. [Google Scholar] [CrossRef] [Green Version]
- Xu, T.; Luo, Y.; Wang, J.; Zhang, N.; Gu, C.; Li, L.; Qian, D.; Cai, W.; Fan, J.; Yin, G.-Y. Exosomal miRNA-128-3p from mesenchymal stem cells of aged rats regulates osteogenesis and bone fracture healing by targeting Smad5. J. Nanobiotechnol. 2020, 18, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Jiao, G.; Ren, S.; Zhang, X.; Li, C.; Wu, W.; Wang, H.; Liu, H.; Zhou, H.; Chen, Y. Exosomes from bone marrow mesenchymal stem cells enhance fracture healing through the promotion of osteogenesis and angiogenesis in a rat model of nonunion. Stem Cell Res. Ther. 2020, 11, 1–15. [Google Scholar] [CrossRef]
- Goshima, J.; Goldberg, V.M.; Caplan, A. The Origin of Bone Formed in Composite Grafts of Porous Calcium Phosphate Ceramic Loaded With Marrow Cells. Clin. Orthop. Relat. Res. 1991, 274. [Google Scholar] [CrossRef]
- Tasso, R.; Fais, F.; Reverberi, D.; Tortelli, F.; Cancedda, R. The recruitment of two consecutive and different waves of host stem/progenitor cells during the development of tissue-engineered bone in a murine model. Biomaterials 2010, 31, 2121–2129. [Google Scholar] [CrossRef] [PubMed]
- Kitaori, T.; Ito, H.; Schwarz, E.M.; Tsutsumi, R.; Yoshitomi, H.; Oishi, S.; Nakano, M.; Fujii, N.; Nagasawa, T.; Nakamura, T. Stromal cell-derived factor 1/cxcr4 signaling is critical for the recruitment of mesenchymal stem cells to the fracture site during skeletal repair in a mouse model. Arthritis Rheum 2009, 60, 813–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, H. Chemokines in mesenchymal stem cell therapy for bone repair: A novel concept of recruiting mesenchymal stem cells and the possible cell sources. Mod. Rheumatol. 2011, 21, 113–121. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, K.R.; Stutz, C.M.; Mignemi, N.A.; Burns, M.C.; Murry, M.R.; Nyman, J.S.; Schoenecker, J.G. Micro-computed tomography assessment of the progression of fracture healing in mice. Bone 2012, 50, 1357–1367. [Google Scholar] [CrossRef]
- Cozen, L. Does diabetes delay fracture healing? Clin. Orthop. Relat. Res. 1972, 82, 134–140. [Google Scholar] [CrossRef]
- Ko, K.I.; Coimbra, L.S.; Tian, C.; Alblowi, J.; Kayal, R.A.; Einhorn, T.; Gerstenfeld, L.C.; Pignolo, R.J.; Graves, D.T. Diabetes reduces mesenchymal stem cells in fracture healing through a TNFα-mediated mechanism. Diabetologia 2015, 58, 633–642. [Google Scholar] [CrossRef] [Green Version]
- Tyndall, W.A.; Beam, H.A.; Zarro, C.; O???connor, J.P.; Lin, S.S. Decreased Platelet Derived Growth Factor Expression During Fracture Healing in Diabetic Animals. Clin. Orthop. Relat. Res. 2003, 408, 319–330. [Google Scholar] [CrossRef]
- Beam, H.A.; Parsons, J.R.; Lin, S.S. The effects of blood glucose control upon fracture healing in the BB Wistar rat with diabetes mellitus. J. Orthop. Res. 2002, 20, 1210–1216. [Google Scholar] [CrossRef]
- Gooch, H.L.; Hale, J.E.; Fujioka, H.; Balian, G.; Hurwitz, S.R. Alterations of cartilage and collagen expression during fracture healing in experimental diabetes. Connect. Tissue Res. 2000, 41, 81–91. [Google Scholar] [CrossRef]
- Xu, M.; Sun, S.; Zhang, L.; Xu, F.; Du, S.; Zhang, X.; Wang, D. Diabetes mellitus affects the biomechanical function of the callus and the expression of TGF-beta1 and BMP2 in an early stage of fracture healing. Braz. J. Med. Boil. Res. 2016, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herbsman, H.; Powers, J.C.; Hirschman, A.; Shaftan, G.W. Retardation of fracture healing in experimental diabetes. J. Surg. Res. 1968, 8, 424–431. [Google Scholar] [CrossRef]
- Diniz, S.; Amorim, F.; Cavalcante-Neto, F.; Bocca, A.L.; Batista, A.C.; Simm, G.; Da Silva, T.A. Alloxan-induced diabetes delays repair in a rat model of closed tibial fracture. Braz. J. Med. Boil. Res. 2008, 41, 373–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dienelt, A.; Nieden, N.I.Z. Hyperglycemia Impairs Skeletogenesis from Embryonic Stem Cells by Affecting Osteoblast and Osteoclast Differentiation. Stem Cells Dev. 2011, 20, 465–474. [Google Scholar] [CrossRef] [PubMed]
- Kasahara, T.; Imai, S.; Kojima, H.; Katagi, M.; Kimura, H.; Chan, L.; Matsusue, Y. Malfunction of bone marrow-derived osteoclasts and the delay of bone fracture healing in diabetic mice. Bone 2010, 47, 617–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wittrant, Y.; Gorin, Y.; Woodruff, K.; Horn, D.; Abboud, H.; Mohan, S.; Abboud-Werner, S. High d(+)glucose concentration inhibits RANKL-induced osteoclastogenesis. Bone 2008, 42, 1122–1130. [Google Scholar] [CrossRef] [Green Version]
- Currey, J.D.; Yonge, C.M. Effects of differences in mineralization on the mechanical properties of bone. Philos. Trans. R. Soc. B: Boil. Sci. 1984, 304, 509–518. [Google Scholar] [CrossRef]
- Ruffoni, D.; Fratzl, P.; Roschger, P.; Klaushofer, K.; Weinkamer, R. The bone mineralization density distribution as a fingerprint of the mineralization process. Bone 2007, 40, 1308–1319. [Google Scholar] [CrossRef]
- GE, V.; AL, K. Bone strength - its relationship to x-ray-determined ash content. Human Biology 1959, 31, 261–270. [Google Scholar]
- Brennan, M.Á.; Gleeson, J.P.; Browne, M.; O’Brien, F.J.; Thurner, P.J.; McNamara, L.M. Site specific increase in heterogeneity of trabecular bone tissue mineral during oestrogen deficiency. Eur Cell Mater. 2011, 21, 396–406. [Google Scholar] [CrossRef]
- Hou, J.C.-H.; Zernicke, R.F.; Barnard, R.J. Experimental Diabetes, Insulin Treatment, and Femoral Neck Morphology and Biomechanics in Rats. Clin. Orthop. Relat. Res. 1991, 264, 278–285. [Google Scholar] [CrossRef]
- Reddy, G.K.; Stehno-Bittel, L.; Hamade, S.; Enwemeka, C.S. The biomechanical integrity of bone in experimental diabetes. Diabetes Res. Clin. Pr. 2001, 54, 1–8. [Google Scholar] [CrossRef]
- Korres, N.; Tsiridis, E.; Pavlou, G.; Mitsoudis, A.; Perrea, D.N.; Zoumbos, A.B. Biomechanical characteristics of bone in streptozotocin-induced diabetic rats: An in-vivo randomized controlled experimental study. World J. Orthop. 2013, 4, 124–129. [Google Scholar] [CrossRef]
- Nyman, J.S.; Even, J.L.; Jo, C.-H.; Herbert, E.G.; Murry, M.R.; Cockrell, G.E.; Wahl, E.C.; Bunn, R.C.; Lumpkin, C.K.; Fowlkes, J.L.; et al. Increasing duration of type 1 diabetes perturbs the strength–structure relationship and increases brittleness of bone. Bone 2011, 48, 733–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Funk, J.R.; Hale, J.E.; Carmines, D.; Gooch, H.L.; Hurwitz, S.R. Biomechanical evaluation of early fracture healing in normal and diabetic rats. J Orthop Res 2000, 18, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Lanier, L.L.; O’Fallon, S.; Somoza, C.; Phillips, J.H.; Linsley, P.S.; Okumura, K.; Ito, D.; Azuma, M. CD80 (B7) and CD86 (B70) provide similar costimulatory signals for T cell proliferation, cytokine production, and generation of CTL. J. Immunol. 1995, 154, 97–105. [Google Scholar]
- Grau-Vorster, M.; Laitinen, A.; Nystedt, J.; Vives, J. HLA-DR expression in clinical-grade bone marrow-derived multipotent mesenchymal stromal cells: A two-site study. Stem Cell Res. Ther. 2019, 10, 164. [Google Scholar] [CrossRef] [Green Version]
- Bocelli-Tyndall, C.; Zajac, P.; Di Maggio, N.; Trella, E.; Benvenuto, F.; Iezzi, G.; Scherberich, A.; Barbero, A.; Schaeren, S.; Pistoia, V.; et al. Fibroblast growth factor 2 and platelet-derived growth factor, but not platelet lysate, induce proliferation-dependent, functional class II major histocompatibility complex antigen in human mesenchymal stem cells. Arthritis Rheum. 2010, 62, 3815–3825. [Google Scholar] [CrossRef]
- Lohan, P.; Treacy, O.; Morcos, M.; Donohoe, E.; O’Donoghue, Y.; Ryan, A.; Elliman, S.J.; Ritter, T.; Griffin, M.D. Interspecies Incompatibilities Limit the Immunomodulatory Effect of Human Mesenchymal Stromal Cells in the Rat. STEM CELLS 2018, 36, 1210–1215. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Small Intestine | Spleen | Muscle | Large Intestine | Liver | Stomach | Pancreas | Kidney | Lung | Bone | Heart | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Day 0 | ND | ND | 0.07 | ND | ND | ND | ND | ND | ND | 0.01 | ND |
| ND | ND | ND | ND | ND | ND | ND | ND | ND | 253 | ND | |
| ND | ND | 365 | ND | ND | ND | ND | ND | ND | ND | ND | |
| ND | ND | 1258 | ND | ND | ND | ND | ND | ND | ND | ND | |
| Day 1 | ND | ND | 4450 | ND | ND | ND | ND | ND | ND | 7 | ND |
| ND | ND | 3165 | ND | ND | ND | ND | ND | ND | 3 | ND | |
| ND | ND | 22,082 | ND | ND | ND | ND | ND | ND | 53 | 6 | |
| ND | ND | 68 | 19 | ND | ND | ND | ND | ND | 165 | ND | |
| Day 2 | ND | ND | 4512 | ND | ND | ND | ND | 158 | ND | ND | ND |
| ND | ND | 5082 | ND | ND | ND | ND | ND | ND | ND | ND | |
| ND | ND | 2905 | ND | ND | ND | ND | ND | ND | 6 | ND | |
| ND | ND | 2595 | ND | ND | ND | ND | ND | ND | ND | ND | |
| ND | ND | 1378 | ND | ND | ND | ND | ND | ND | ND | ND | |
| Day 3 | ND | ND | 3510 | ND | ND | ND | ND | ND | ND | ND | ND |
| ND | ND | 495 | ND | ND | ND | ND | ND | ND | ND | ND | |
| ND | ND | 416 | ND | ND | ND | ND | ND | ND | 13 | ND | |
| ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | |
| ND | ND | 149 | ND | ND | ND | ND | ND | ND | 190 | ND | |
| Day 7 | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND |
| ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | |
| ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | |
| ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | |
| ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | |
| Negative Control | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND |
| ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | |
| ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | |
| ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Watson, L.; Chen, X.Z.; Ryan, A.E.; Fleming, Á.; Carbin, A.; O’Flynn, L.; Loftus, P.G.; Horan, E.; Connolly, D.; McDonnell, P.; et al. Administration of Human Non-Diabetic Mesenchymal Stromal Cells to a Murine Model of Diabetic Fracture Repair: A Pilot Study. Cells 2020, 9, 1394. https://doi.org/10.3390/cells9061394
Watson L, Chen XZ, Ryan AE, Fleming Á, Carbin A, O’Flynn L, Loftus PG, Horan E, Connolly D, McDonnell P, et al. Administration of Human Non-Diabetic Mesenchymal Stromal Cells to a Murine Model of Diabetic Fracture Repair: A Pilot Study. Cells. 2020; 9(6):1394. https://doi.org/10.3390/cells9061394
Chicago/Turabian StyleWatson, Luke, Xi Zhe Chen, Aideen E. Ryan, Áine Fleming, Aoife Carbin, Lisa O’Flynn, Paul G. Loftus, Emma Horan, David Connolly, Patrick McDonnell, and et al. 2020. "Administration of Human Non-Diabetic Mesenchymal Stromal Cells to a Murine Model of Diabetic Fracture Repair: A Pilot Study" Cells 9, no. 6: 1394. https://doi.org/10.3390/cells9061394
APA StyleWatson, L., Chen, X. Z., Ryan, A. E., Fleming, Á., Carbin, A., O’Flynn, L., Loftus, P. G., Horan, E., Connolly, D., McDonnell, P., McNamara, L. M., O’Brien, T., & Coleman, C. M. (2020). Administration of Human Non-Diabetic Mesenchymal Stromal Cells to a Murine Model of Diabetic Fracture Repair: A Pilot Study. Cells, 9(6), 1394. https://doi.org/10.3390/cells9061394