Exploring Therapeutic Targets to Reverse or Prevent the Transition from Metabolically Healthy to Unhealthy Obesity


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Obesity: A Heterogeneous Disorder of Rapidly Increasing Global Importance
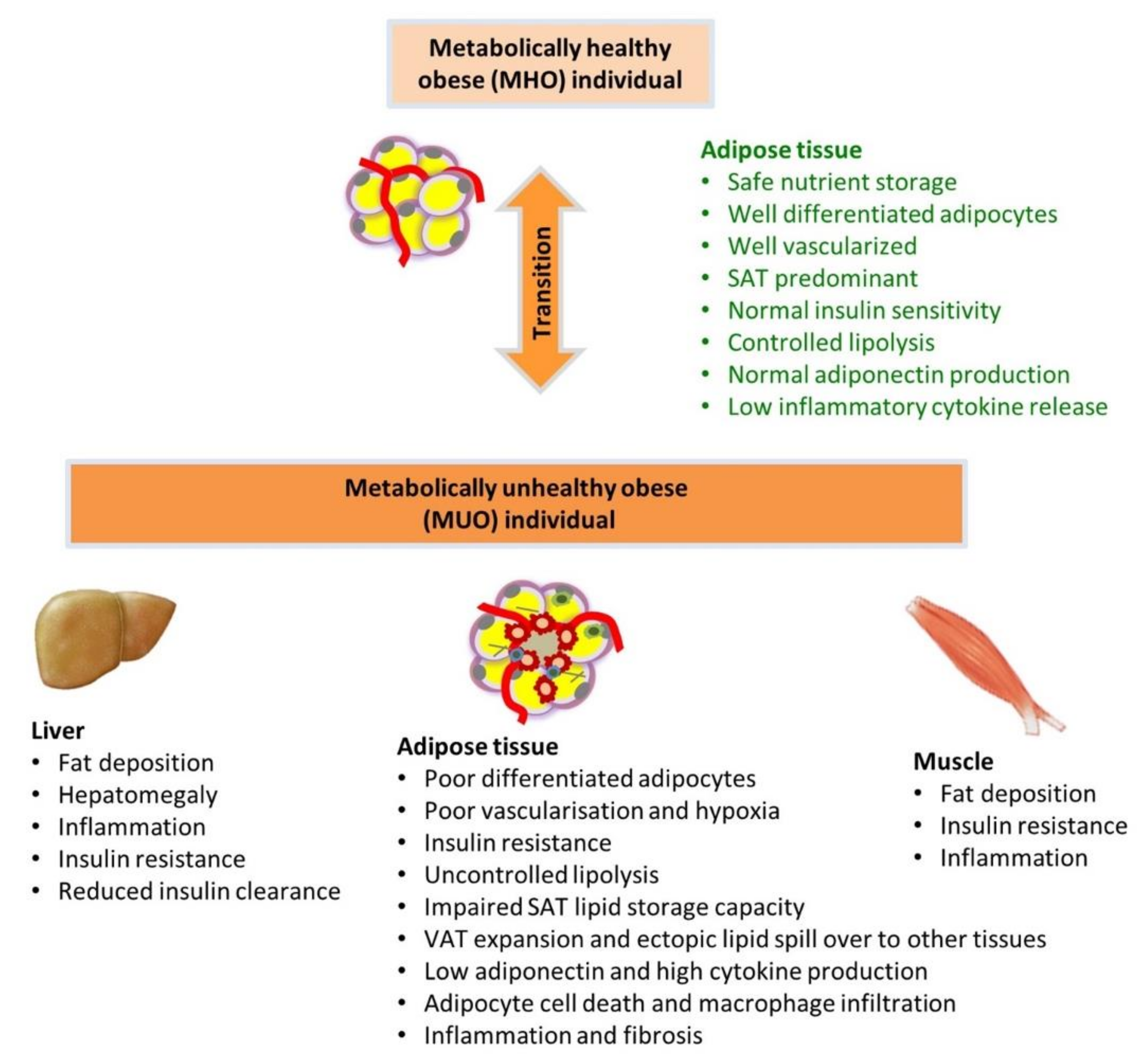
2. Classification of Obesity as Metabolically Healthy or Unhealthy
3. Adipose Tissue Types, Function and Sites
4. Healthy and Unhealthy Responses of Adipose Tissue to Excessive Nutrient Supply
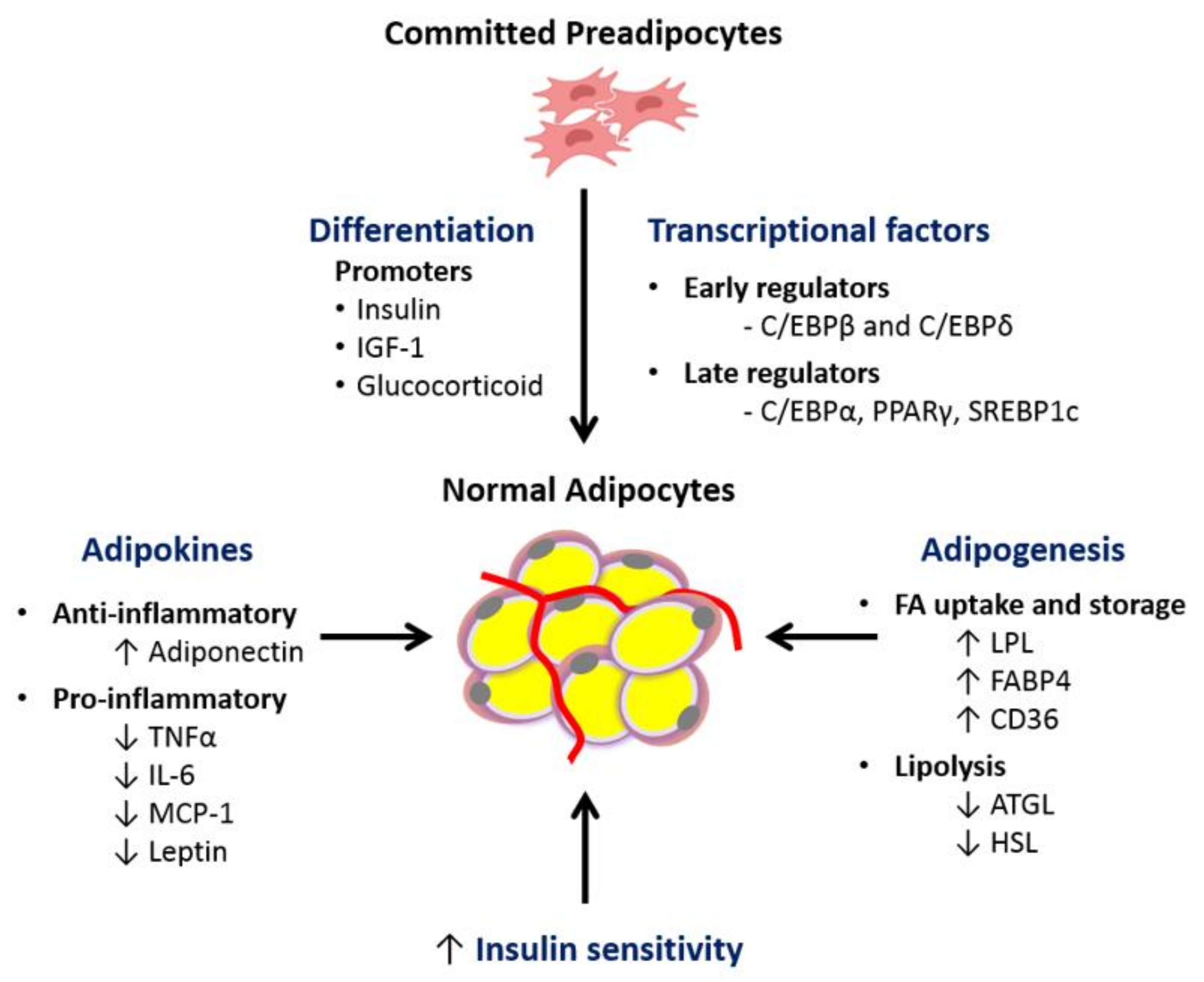
4.1. Impaired Adipogenesis and Adipocyte Differentiation Capacity
4.2. Hypoxia-Induced Adipose Tissue Injury and Adipose Tissue Inflammation
4.3. Altered Adipokine/Cytokine Production
4.4. Dysfunctional Adipose Tissue Lipid Metabolism
4.5. Role of Epigenetics
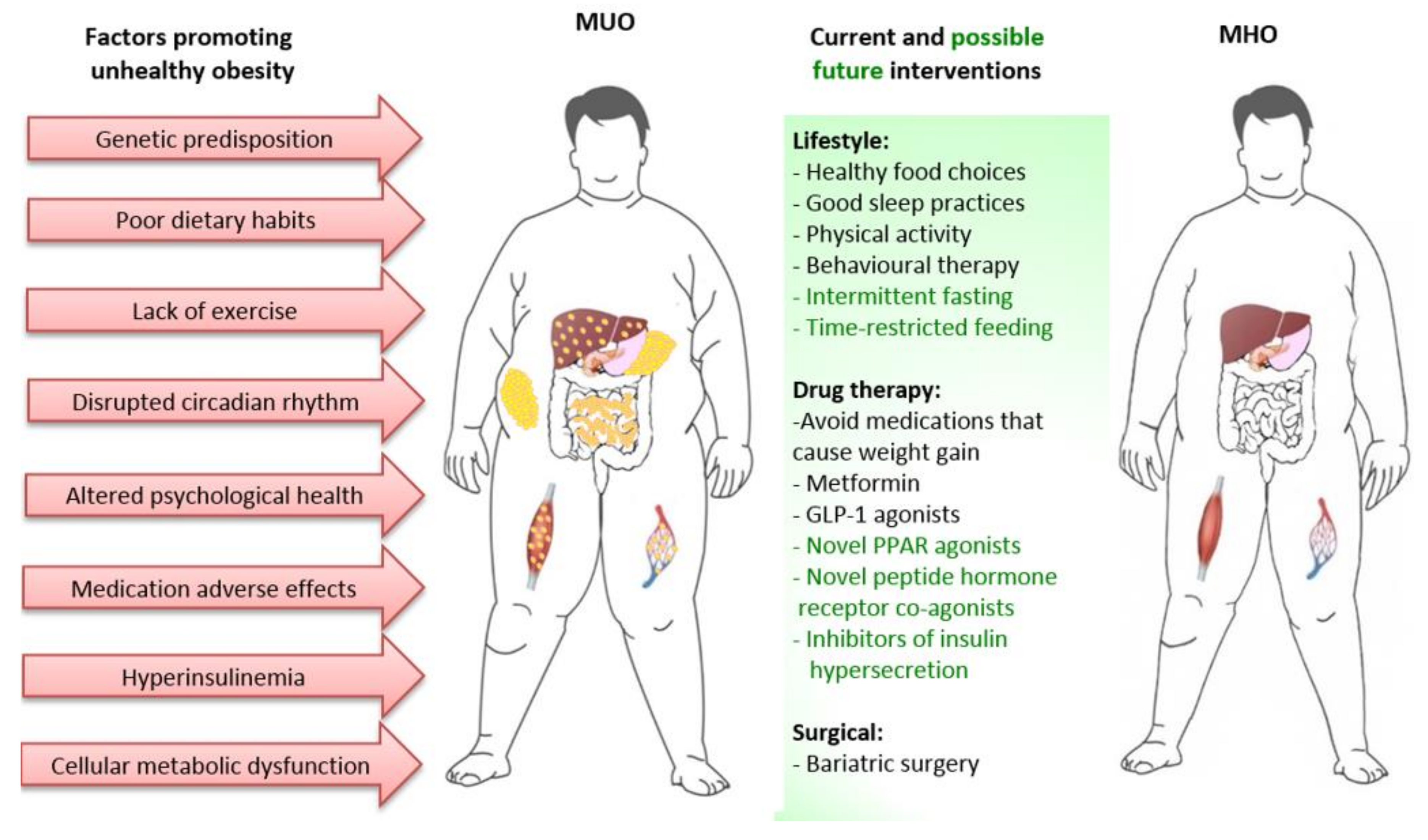
5. Factors Extrinsic to Adipose Tissue Contributing to MUO
5.1. Total Body Energy Balance, Pattern of Eating, Nutrient Quality, and Exercise
5.2. Disruptions in the Circadian Rhythm
5.3. Potential Role of Hyperinsulinemia as Driver to MUO Phenotype
5.4. Altered Whole Body Amino Acid Metabolism
6. Approaches to Prevent or Reverse the Progression from MHO to MUO
6.1. Predicting Subjects at Risk of Progression to MUO
6.2. Lifestyle Interventions with no or Minimal Weight Loss
6.3. Pharmaceutical Interventions with no or Minimal Weight Loss
6.4. Pharmaceutical Interventions with greater effect on Weight Loss
6.5. Bariatric Surgery
6.6. Preventing Insulin Hypersecretion
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kopelman, P.G. Obesity as a medical problem. Nature 2000, 404, 635–643. [Google Scholar] [CrossRef] [PubMed]
- Ng, M.; Fleming, T.; Robinson, M.; Thomson, B.; Graetz, N.; Margono, C.; Mullany, E.C.; Biryukov, S.; Abbafati, C.; Abera, S.F.; et al. Global, regional, and national prevalence of overweight and obesity in children and adults during 1980-2013: A systematic analysis for the Global Burden of Disease Study 2013. Lancet 2014, 384, 766–781. [Google Scholar] [CrossRef] [Green Version]
- El-Sayed Moustafa, J.S.; Froguel, P. From obesity genetics to the future of personalized obesity therapy. Nat. Rev. Endocrinol. 2013, 9, 402–413. [Google Scholar] [CrossRef] [PubMed]
- Frayling, T.M.; Timpson, N.J.; Weedon, M.N.; Zeggini, E.; Freathy, R.M.; Lindgren, C.M.; Perry, J.R.; Elliott, K.S.; Lango, H.; Rayner, N.W.; et al. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science 2007, 316, 889–894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navarro, E.; Funtikova, A.N.; Fíto, M.; Schröder, H. Can Metabolically Healthy Obesity be explained by diet, genetics and inflammation? Mol. Nutr. Food Res. 2014, 59, 75–93. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027–1131. [Google Scholar] [CrossRef]
- Metzger, B.E.; Lowe, L.P.; Dyer, A.R.; Trimble, E.R.; Chaovarindr, U.; Coustan, D.R.; Hadden, D.R.; McCance, D.R.; Hod, M.; McIntyre, H.D.; et al. Hyperglycemia and adverse pregnancy outcomes. N. Engl. J. Med. 2008, 358, 1991–2002. [Google Scholar] [CrossRef] [Green Version]
- Poston, L.; Caleyachetty, R.; Cnattingius, S.; Corvalan, C.; Uauy, R.; Herring, S.; Gillman, M.W. Preconceptional and maternal obesity: Epidemiology and health consequences. Lancet Diabetes Endocrinol. 2016, 4, 1025–1036. [Google Scholar] [CrossRef]
- Baugh, N.; Harris, D.E. The Impact of Maternal Obesity and Excessive Gestational Weight Gain on Maternal and Infant Outcomes in Maine: Analysis of Pregnancy Risk Assessment Monitoring System Results from 2000 to 2010. J. Pregnancy 2016, 2016, 5871313. [Google Scholar] [CrossRef] [Green Version]
- Gluckman, P.D.; Hanson, M.; Zimmet, P.; Forrester, T. Losing the war against obesity: The need for a developmental perspective. Sci. Transl. Med. 2011, 3, 93cm19. [Google Scholar] [CrossRef] [Green Version]
- Afshin, A.; Forouzanfar, M.H.; Reitsma, M.B.; Sur, P.; Estep, K.; Lee, A.; Marczak, L.; Mokdad, A.H.; Moradi-Lakeh, M.; et al.; GBD Obesity Collaborators Health Effects of Overweight and Obesity in 195 Countries over 25 Years. N. Engl. J. Med. 2017, 377, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Bray, G.A. Medical consequences of obesity. J. Clin. Endocrinol. Metab. 2004, 89, 2583–2589. [Google Scholar] [CrossRef] [Green Version]
- Renehan, A.G.; Zwahlen, M.; Egger, M. Adiposity and cancer risk: New mechanistic insights from epidemiology. Nat. Rev. Cancer 2015, 15, 484–498. [Google Scholar] [CrossRef] [PubMed]
- Malvi, P.; Chaube, B.; Singh, S.V.; Mohammad, N.; Pandey, V.; Vijayakumar, M.V.; Radhakrishnan, R.M.; Vanuopadath, M.; Nair, S.S.; Nair, B.G.; et al. Weight control interventions improve therapeutic efficacy of dacarbazine in melanoma by reversing obesity-induced drug resistance. Cancer Metab. 2016, 4, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nolan, C.J.; Prentki, M. Insulin resistance and insulin hypersecretion in the metabolic syndrome and type 2 diabetes: Time for a conceptual framework shift. Diab. Vasc. Dis. Res. 2019. [Google Scholar] [CrossRef] [PubMed]
- Reaven, G.M. Banting lecture 1988. Role of insulin resistance in human disease. Diabetes 1988, 37, 1595–1607. [Google Scholar] [CrossRef]
- Withrow, D.; Alter, D.A. The economic burden of obesity worldwide: A systematic review of the direct costs of obesity. Obes. Rev. 2011, 12, 131–141. [Google Scholar] [CrossRef]
- Heymsfield, S.B.; Wadden, T.A. Mechanisms, Pathophysiology, and Management of Obesity. N. Engl. J. Med. 2017, 376, 254–266. [Google Scholar] [CrossRef]
- Smith, G.I.; Mittendorfer, B.; Klein, S. Metabolically healthy obesity: Facts and fantasies. J. Clin. Invest. 2019, 129, 3978–3989. [Google Scholar] [CrossRef] [Green Version]
- Wildman, R.P.; Muntner, P.; Reynolds, K.; McGinn, A.P.; Rajpathak, S.; Wylie-Rosett, J.; Sowers, M.R. The obese without cardiometabolic risk factor clustering and the normal weight with cardiometabolic risk factor clustering: Prevalence and correlates of 2 phenotypes among the US population (NHANES 1999-2004). Arch. Intern. Med. 2008, 168, 1617–1624. [Google Scholar] [CrossRef] [Green Version]
- Primeau, V.; Coderre, L.; Karelis, A.D.; Brochu, M.; Lavoie, M.E.; Messier, V.; Sladek, R.; Rabasa-Lhoret, R. Characterizing the profile of obese patients who are metabolically healthy. Int. J. Obes. 2011, 35, 971–981. [Google Scholar] [CrossRef] [Green Version]
- Schroder, H.; Ramos, R.; Baena-Diez, J.M.; Mendez, M.A.; Canal, D.J.; Fito, M.; Sala, J.; Elosua, R. Determinants of the transition from a cardiometabolic normal to abnormal overweight/obese phenotype in a Spanish population. Eur. J. Nutr. 2014, 53, 1345–1353. [Google Scholar] [CrossRef] [PubMed]
- Kramer, C.K.; Zinman, B.; Retnakaran, R. Are metabolically healthy overweight and obesity benign conditions?: A systematic review and meta-analysis. Ann. Intern. Med. 2013, 159, 758–769. [Google Scholar] [CrossRef]
- Appleton, S.L.; Seaborn, C.J.; Visvanathan, R.; Hill, C.L.; Gill, T.K.; Taylor, A.W.; Adams, R.J. Diabetes and cardiovascular disease outcomes in the metabolically healthy obese phenotype: A cohort study. Diabetes Care 2013, 36, 2388–2394. [Google Scholar] [CrossRef] [Green Version]
- Hamer, M.; Bell, J.A.; Sabia, S.; Batty, G.D.; Kivimaki, M. Stability of metabolically healthy obesity over 8 years: The English Longitudinal Study of Ageing. Eur. J. Endocrinol. 2015, 173, 703–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eshtiaghi, R.; Keihani, S.; Hosseinpanah, F.; Barzin, M.; Azizi, F. Natural course of metabolically healthy abdominal obese adults after 10 years of follow-up: The Tehran Lipid and Glucose Study. Int. J. Obes. 2015, 39, 514–519. [Google Scholar] [CrossRef] [PubMed]
- Cinti, S. Anatomy and physiology of the nutritional system. Mol. Asp. Med. 2019, 68, 101–107. [Google Scholar] [CrossRef]
- Iozzo, P. Myocardial, perivascular, and epicardial fat. Diabetes Care 2011, 34 Suppl 2, S371–379. [Google Scholar] [CrossRef] [Green Version]
- Bartelt, A.; Heeren, J. Adipose tissue browning and metabolic health. Nat. Rev. Endocrinol. 2014, 10, 24–36. [Google Scholar] [CrossRef]
- Morroni, M.; Giordano, A.; Zingaretti, M.C.; Boiani, R.; De Matteis, R.; Kahn, B.B.; Nisoli, E.; Tonello, C.; Pisoschi, C.; Luchetti, M.M.; et al. Reversible transdifferentiation of secretory epithelial cells into adipocytes in the mammary gland. Proc. Natl. Acad. Sci. USA 2004, 101, 16801–16806. [Google Scholar] [CrossRef] [Green Version]
- Prokesch, A.; Smorlesi, A.; Perugini, J.; Manieri, M.; Ciarmela, P.; Mondini, E.; Trajanoski, Z.; Kristiansen, K.; Giordano, A.; Bogner-Strauss, J.G.; et al. Molecular aspects of adipoepithelial transdifferentiation in mouse mammary gland. Stem Cells. 2014, 32, 2756–2766. [Google Scholar] [CrossRef] [PubMed]
- Méndez-Sánchez, N.; Arrese, M.; Zamora-Valdés, D.; Uribe, M. Current concepts in the pathogenesis of nonalcoholic fatty liver disease. Liver. Int. 2007, 27, 423–433. [Google Scholar] [CrossRef]
- Oliva-Olivera, W.; Moreno-Indias, I.; Coin-Araguez, L.; Lhamyani, S.; Alcaide Torres, J.; Fernandez-Veledo, S.; Vendrell, J.; Camargo, A.; El Bekay, R.; Tinahones, F.J. Different response to hypoxia of adipose-derived multipotent cells from obese subjects with and without metabolic syndrome. PLoS ONE 2017, 12, e0188324. [Google Scholar] [CrossRef] [Green Version]
- Cottam, D.R.; Mattar, S.G.; Barinas-Mitchell, E.; Eid, G.; Kuller, L.; Kelley, D.E.; Schauer, P.R. The chronic inflammatory hypothesis for the morbidity associated with morbid obesity: Implications and effects of weight loss. Obes. Surg. 2004, 14, 589–600. [Google Scholar] [CrossRef] [PubMed]
- Crewe, C.; An, Y.A.; Scherer, P.E. The ominous triad of adipose tissue dysfunction: Inflammation, fibrosis, and impaired angiogenesis. J. Clin. Invest. 2017, 127, 74–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otto, T.C.; Lane, M.D. Adipose development: From stem cell to adipocyte. Crit. Rev. Biochem. Mol. Biol. 2005, 40, 229–242. [Google Scholar] [CrossRef]
- Wang, Q.A.; Tao, C.; Gupta, R.K.; Scherer, P.E. Tracking adipogenesis during white adipose tissue development, expansion and regeneration. Nat. Med. 2013, 19, 1338–1344. [Google Scholar] [CrossRef]
- Ntambi, J.M.; Young-Cheul, K. Adipocyte differentiation and gene expression. J. Nutr. 2000, 130, 3122s–3126s. [Google Scholar] [CrossRef]
- Lefterova, M.I.; Lazar, M.A. New developments in adipogenesis. Trends Endocrinol. Metab. 2009, 20, 107–114. [Google Scholar] [CrossRef]
- Telle-Hansen, V.H.; Halvorsen, B.; Dalen, K.T.; Narverud, I.; Wesseltoft-Rao, N.; Granlund, L.; Ulven, S.M.; Holven, K.B. Altered expression of genes involved in lipid metabolism in obese subjects with unfavourable phenotype. Genes Nutr. 2013, 8, 425–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLaughlin, T.; Sherman, A.; Tsao, P.; Gonzalez, O.; Yee, G.; Lamendola, C.; Reaven, G.M.; Cushman, S.W. Enhanced proportion of small adipose cells in insulin-resistant vs insulin-sensitive obese individuals implicates impaired adipogenesis. Diabetologia 2007, 50, 1707–1715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tinahones, F.J.; Garrido-Sanchez, L.; Miranda, M.; Garcia-Almeida, J.M.; Macias-Gonzalez, M.; Ceperuelo, V.; Gluckmann, E.; Rivas-Marin, J.; Vendrell, J.; Garcia-Fuentes, E. Obesity and insulin resistance-related changes in the expression of lipogenic and lipolytic genes in morbidly obese subjects. Obes. Surg. 2010, 20, 1559–1567. [Google Scholar] [CrossRef] [PubMed]
- Tchkonia, T.; Lenburg, M.; Thomou, T.; Giorgadze, N.; Frampton, G.; Pirtskhalava, T.; Cartwright, A.; Cartwright, M.; Flanagan, J.; Karagiannides, I.; et al. Identification of depot-specific human fat cell progenitors through distinct expression profiles and developmental gene patterns. Am. J. Physiol. Endocrinol. Metab. 2007, 292, E298–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, L.; Guo, F.; Zhou, L.; Stahl, R.; Grams, J. The cell size and distribution of adipocytes from subcutaneous and visceral fat is associated with type 2 diabetes mellitus in humans. Adipocyte 2015, 4, 273–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLaughlin, T.; Lamendola, C.; Coghlan, N.; Liu, T.C.; Lerner, K.; Sherman, A.; Cushman, S.W. Subcutaneous adipose cell size and distribution: Relationship to insulin resistance and body fat. Obesity 2014, 22, 673–680. [Google Scholar] [CrossRef] [Green Version]
- Verboven, K.; Wouters, K.; Gaens, K.; Hansen, D.; Bijnen, M.; Wetzels, S.; Stehouwer, C.D.; Goossens, G.H.; Schalkwijk, C.G.; Blaak, E.E.; et al. Abdominal subcutaneous and visceral adipocyte size, lipolysis and inflammation relate to insulin resistance in male obese humans. Sci. Rep. 2018, 8, 4677. [Google Scholar] [CrossRef]
- Belligoli, A.; Compagnin, C.; Sanna, M.; Favaretto, F.; Fabris, R.; Busetto, L.; Foletto, M.; Dal Pra, C.; Serra, R.; Prevedello, L.; et al. Characterization of subcutaneous and omental adipose tissue in patients with obesity and with different degrees of glucose impairment. Sci. Rep. 2019, 9, 11333. [Google Scholar] [CrossRef]
- Lackey, D.E.; Burk, D.H.; Ali, M.R.; Mostaedi, R.; Smith, W.H.; Park, J.; Scherer, P.E.; Seay, S.A.; McCoin, C.S.; Bonaldo, P.; et al. Contributions of adipose tissue architectural and tensile properties toward defining healthy and unhealthy obesity. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E233–246. [Google Scholar] [CrossRef] [Green Version]
- He, W.; Barak, Y.; Hevener, A.; Olson, P.; Liao, D.; Le, J.; Nelson, M.; Ong, E.; Olefsky, J.M.; Evans, R.M. Adipose-specific peroxisome proliferator-activated receptor gamma knockout causes insulin resistance in fat and liver but not in muscle. Proc. Natl. Acad. Sci. USA 2003, 100, 15712–15717. [Google Scholar] [CrossRef] [Green Version]
- Sugii, S.; Olson, P.; Sears, D.D.; Saberi, M.; Atkins, A.R.; Barish, G.D.; Hong, S.H.; Castro, G.L.; Yin, Y.Q.; Nelson, M.C.; et al. PPARgamma activation in adipocytes is sufficient for systemic insulin sensitization. Proc. Natl. Acad. Sci. USA 2009, 106, 22504–22509. [Google Scholar] [CrossRef] [Green Version]
- Maeda, N.; Takahashi, M.; Funahashi, T.; Kihara, S.; Nishizawa, H.; Kishida, K.; Nagaretani, H.; Matsuda, M.; Komuro, R.; Ouchi, N.; et al. PPARgamma ligands increase expression and plasma concentrations of adiponectin, an adipose-derived protein. Diabetes 2001, 50, 2094–2099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.B.; Wright, H.M.; Wright, M.; Spiegelman, B.M. ADD1/SREBP1 activates PPARgamma through the production of endogenous ligand. Proc. Natl. Acad. Sci. USA 1998, 95, 4333–4337. [Google Scholar] [PubMed] [Green Version]
- Wernstedt Asterholm, I.; Tao, C.; Morley, T.S.; Wang, Q.A.; Delgado-Lopez, F.; Wang, Z.V.; Scherer, P.E. Adipocyte inflammation is essential for healthy adipose tissue expansion and remodeling. Cell Metab. 2014, 20, 103–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strissel, K.J.; Stancheva, Z.; Miyoshi, H.; Perfield, J.W., 2nd; DeFuria, J.; Jick, Z.; Greenberg, A.S.; Obin, M.S. Adipocyte death, adipose tissue remodeling, and obesity complications. Diabetes 2007, 56, 2910–2918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, T.; Muise, E.S.; Iyengar, P.; Wang, Z.V.; Chandalia, M.; Abate, N.; Zhang, B.B.; Bonaldo, P.; Chua, S.; Scherer, P.E. Metabolic dysregulation and adipose tissue fibrosis: Role of collagen VI. Mol. Cell Biol. 2009, 29, 1575–1591. [Google Scholar] [CrossRef] [Green Version]
- Koumenis, C.; Naczki, C.; Koritzinsky, M.; Rastani, S.; Diehl, A.; Sonenberg, N.; Koromilas, A.; Wouters, B.G. Regulation of protein synthesis by hypoxia via activation of the endoplasmic reticulum kinase PERK and phosphorylation of the translation initiation factor eIF2alpha. Mol. Cell Biol. 2002, 22, 7405–7416. [Google Scholar] [CrossRef] [Green Version]
- Halberg, N.; Khan, T.; Trujillo, M.E.; Wernstedt-Asterholm, I.; Attie, A.D.; Sherwani, S.; Wang, Z.V.; Landskroner-Eiger, S.; Dineen, S.; Magalang, U.J.; et al. Hypoxia-inducible factor 1alpha induces fibrosis and insulin resistance in white adipose tissue. Mol. Cell Biol. 2009, 29, 4467–4483. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.S.; Kim, J.W.; Osborne, O.; Oh, D.Y.; Sasik, R.; Schenk, S.; Chen, A.; Chung, H.; Murphy, A.; Watkins, S.M.; et al. Increased adipocyte O2 consumption triggers HIF-1alpha, causing inflammation and insulin resistance in obesity. Cell 2014, 157, 1339–1352. [Google Scholar] [CrossRef] [Green Version]
- Petrus, P.; Fernandez, T.L.; Kwon, M.M.; Huang, J.L.; Lei, V.; Safikhan, N.S.; Karunakaran, S.; O’Shannessy, D.J.; Zheng, X.; Catrina, S.B.; et al. Specific loss of adipocyte CD248 improves metabolic health via reduced white adipose tissue hypoxia, fibrosis and inflammation. EBioMedicine 2019, 44, 489–501. [Google Scholar] [CrossRef] [Green Version]
- Bluher, M.; Mantzoros, C.S. From leptin to other adipokines in health and disease: Facts and expectations at the beginning of the 21st century. Metabolism 2015, 64, 131–145. [Google Scholar] [CrossRef]
- Arita, Y.; Kihara, S.; Ouchi, N.; Takahashi, M.; Maeda, K.; Miyagawa, J.; Hotta, K.; Shimomura, I.; Nakamura, T.; Miyaoka, K.; et al. Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. Biochem. Biophys. Res. Commun. 1999, 257, 79–83. [Google Scholar] [PubMed]
- Kwon, H.; Pessin, J.E. Adipokines Mediate Inflammation and Insulin Resistance. Front. Endocrinol. 2013, 4, 71. [Google Scholar] [CrossRef] [Green Version]
- Cao, H. Adipocytokines in obesity and metabolic disease. J. Endocrinol. 2014, 220, T47–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamauchi, T.; Kamon, J.; Minokoshi, Y.; Ito, Y.; Waki, H.; Uchida, S.; Yamashita, S.; Noda, M.; Kita, S.; Ueki, K.; et al. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat. Med. 2002, 8, 1288–1295. [Google Scholar] [CrossRef] [PubMed]
- Kumada, M.; Kihara, S.; Ouchi, N.; Kobayashi, H.; Okamoto, Y.; Ohashi, K.; Maeda, K.; Nagaretani, H.; Kishida, K.; Maeda, N.; et al. Adiponectin specifically increased tissue inhibitor of metalloproteinase-1 through interleukin-10 expression in human macrophages. Circulation 2004, 109, 2046–2049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caro, J.F.; Kolaczynski, J.W.; Nyce, M.R.; Ohannesian, J.P.; Opentanova, I.; Goldman, W.H.; Lynn, R.B.; Zhang, P.L.; Sinha, M.K.; Considine, R.V. Decreased cerebrospinal-fluid/serum leptin ratio in obesity: A possible mechanism for leptin resistance. Lancet 1996, 348, 159–161. [Google Scholar] [CrossRef]
- Kolaczynski, J.W.; Considine, R.V.; Ohannesian, J.; Marco, C.; Opentanova, I.; Nyce, M.R.; Myint, M.; Caro, J.F. Responses of leptin to short-term fasting and refeeding in humans: A link with ketogenesis but not ketones themselves. Diabetes 1996, 45, 1511–1515. [Google Scholar] [CrossRef]
- Fernández-Riejos, P.; Najib, S.; Santos-Alvarez, J.; Martín-Romero, C.; Pérez-Pérez, A.; González-Yanes, C.; Sánchez-Margalet, V. Role of leptin in the activation of immune cells. Mediat. Inflamm. 2010, 2010, 568343. [Google Scholar] [CrossRef]
- Lee, T.H.; Jeon, W.S.; Han, K.J.; Lee, S.Y.; Kim, N.H.; Chae, H.B.; Jang, C.M.; Yoo, K.M.; Park, H.J.; Lee, M.K.; et al. Comparison of Serum Adipocytokine Levels according to Metabolic Health and Obesity Status. Endocrinol. Metab. 2015, 30, 185–194. [Google Scholar] [CrossRef]
- Alfadda, A.A. Circulating Adipokines in Healthy versus Unhealthy Overweight and Obese Subjects. Int. J. Endocrinol. 2014, 2014, 170434. [Google Scholar] [CrossRef]
- Goralski, K.B.; McCarthy, T.C.; Hanniman, E.A.; Zabel, B.A.; Butcher, E.C.; Parlee, S.D.; Muruganandan, S.; Sinal, C.J. Chemerin, a novel adipokine that regulates adipogenesis and adipocyte metabolism. J. Biol. Chem. 2007, 282, 28175–28188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kos, K.; Wilding, J.P. SPARC: A key player in the pathologies associated with obesity and diabetes. Nat. Rev. Endocrinol. 2010, 6, 225–235. [Google Scholar] [CrossRef]
- Ctoi, A.F.; Parvu, A.E.; Andreicut, A.D.; Mironiuc, A.; Crciun, A.; Ctoi, C.; Pop, I.D. Metabolically Healthy versus Unhealthy Morbidly Obese: Chronic Inflammation, Nitro-Oxidative Stress, and Insulin Resistance. Nutrients 2018, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niklowitz, P.; Rothermel, J.; Lass, N.; Barth, A.; Reinehr, T. Link between chemerin, central obesity, and parameters of the Metabolic Syndrome: Findings from a longitudinal study in obese children participating in a lifestyle intervention. Int. J. Obes. 2018, 42, 1743–1752. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Li, Y.; Esangbedo, I.C.; Li, G.; Feng, D.; Li, L.; Xu, L.; Han, L.; Li, M.; Li, C.; et al. Circulating Osteonectin and Adipokine Profiles in Relation to Metabolically Healthy Obesity in Chinese Children: Findings From BCAMS. J. Am. Heart Assoc. 2018, 7, e009169. [Google Scholar] [CrossRef] [Green Version]
- Emanuelli, B.; Peraldi, P.; Filloux, C.; Chavey, C.; Freidinger, K.; Hilton, D.j.; Hotamisligil, G.S.; Van Obberghen, E. SOCS-3 inhibits insulin signaling and is up-regulated in response to tumor necrosis factor-alpha in the adipose tissue of obese mice. J. Biol. Chem. 2001, 276, 47944–47949. [Google Scholar] [CrossRef] [Green Version]
- Klover, P.J.; Zimmers, T.A.; Koniaris, L.G.; Mooney, R.A. Chronic exposure to interleukin-6 causes hepatic insulin resistance in mice. Diabetes 2003, 52, 2784–2789. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Real, J.M.; Broch, M.; Vendrell, J.; Richart, C.; Ricart, W. Interleukin-6 gene polymorphism and lipid abnormalities in healthy subjects. J. Clin. Endocrinol. Metab. 2000, 85, 1334–1339. [Google Scholar] [CrossRef] [Green Version]
- Kanda, H.; Tateya, S.; Tamori, Y.; Kotani, K.; Hiasa, K.; Kitazawa, R.; Kitazawa, S.; Miyachi, H.; Maeda, S.; Egashira, K.; et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J. Clin. Invest. 2006, 116, 1494–1505. [Google Scholar] [CrossRef]
- Shin, M.J.; Hyun, Y.J.; Kim, O.Y.; Kim, J.Y.; Jang, Y.; Lee, J.H. Weight loss effect on inflammation and LDL oxidation in metabolically healthy but obese (MHO) individuals: Low inflammation and LDL oxidation in MHO women. Int. J. Obes. 2006, 30, 1529–1534. [Google Scholar] [CrossRef] [Green Version]
- Phillips, C.M.; Perry, I.J. Does inflammation determine metabolic health status in obese and nonobese adults? J. Clin. Endocrinol. Metab. 2013, 98, E1610–1619. [Google Scholar] [CrossRef] [PubMed]
- Karelis, A.D.; St-Pierre, D.H.; Conus, F.; Rabasa-Lhoret, R.; Poehlman, E.T. Metabolic and Body Composition Factors in Subgroups of Obesity: What Do We Know? J. Clin. Endocrinol. Metab. 2004, 89, 2569–2575. [Google Scholar] [CrossRef]
- Reaven, G. All obese individuals are not created equal: Insulin resistance is the major determinant of cardiovascular disease in overweight/obese individuals. Diab. Vasc. Dis. Res. 2005, 2, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.H.; Huang, J.; Duvel, K.; Boback, B.; Wu, S.; Squillace, R.M.; Wu, C.L.; Manning, B.D. Insulin stimulates adipogenesis through the Akt-TSC2-mTORC1 pathway. PLoS ONE 2009, 4, e6189. [Google Scholar] [CrossRef]
- Semenkovich, C.F.; Wims, M.; Noe, L.; Etienne, J.; Chan, L. Insulin regulation of lipoprotein lipase activity in 3T3-L1 adipocytes is mediated at posttranscriptional and posttranslational levels. J. Biol. Chem. 1989, 264, 9030–9038. [Google Scholar] [PubMed]
- Kern, P.A.; Mandic, A.; Eckel, R.H. Regulation of lipoprotein lipase by glucose in primary cultures of isolated human adipocytes. Relevance to hypertriglyceridemia of diabetes. Diabetes 1987, 36, 1238–1245. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Eckel, R.H. Lipoprotein lipase: From gene to obesity. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E271–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dijk, W.; Kersten, S. Regulation of lipoprotein lipase by Angptl4. Trends Endocrinol. Metab. 2014, 25, 146–155. [Google Scholar] [CrossRef]
- Morin, C.L.; Schlaepfer, I.R.; Eckel, R.H. Tumor necrosis factor-alpha eliminates binding of NF-Y and an octamer-binding protein to the lipoprotein lipase promoter in 3T3-L1 adipocytes. J. Clin. Invest. 1995, 95, 1684–1689. [Google Scholar] [CrossRef] [Green Version]
- Prentki, M.; Madiraju, S.R. Glycerolipid metabolism and signaling in health and disease. Endocr. Rev. 2008, 29, 647–676. [Google Scholar] [CrossRef] [Green Version]
- Lawler, H.M.; Underkofler, C.M.; Kern, P.A.; Erickson, C.; Bredbeck, B.; Rasouli, N. Adipose Tissue Hypoxia, Inflammation, and Fibrosis in Obese Insulin-Sensitive and Obese Insulin-Resistant Subjects. J. Clin. Endocrinol. Metab. 2016, 101, 1422–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lumeng, C.N.; Saltiel, A.R. Inflammatory links between obesity and metabolic disease. J. Clin. Invest. 2011, 121, 2111–2117. [Google Scholar] [CrossRef] [Green Version]
- Bazelmans, J.; Nestel, P.J.; Nolan, C. Insulin-induced glucose utilization influences triglyceride metabolism. Clin. Sci. 1983, 64, 511–516. [Google Scholar] [CrossRef] [PubMed]
- McQuaid, S.E.; Hodson, L.; Neville, M.J.; Dennis, A.L.; Cheeseman, J.; Humphreys, S.M.; Ruge, T.; Gilbert, M.; Fielding, B.A.; Frayn, K.N.; et al. Downregulation of adipose tissue fatty acid trafficking in obesity: A driver for ectopic fat deposition? Diabetes 2011, 60, 47–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auguet, T.; Guiu-Jurado, E.; Berlanga, A.; Terra, X.; Martinez, S.; Porras, J.A.; Ceausu, A.; Sabench, F.; Hernandez, M.; Aguilar, C.; et al. Downregulation of lipogenesis and fatty acid oxidation in the subcutaneous adipose tissue of morbidly obese women. Obesity 2014, 22, 2032–2038. [Google Scholar] [CrossRef] [PubMed]
- Clemente-Postigo, M.; Queipo-Ortuño, M.I.; Fernandez-Garcia, D.; Gomez-Huelgas, R.; Tinahones, F.J.; Cardona, F. Adipose Tissue Gene Expression of Factors Related to Lipid Processing in Obesity. PLoS ONE 2011, 6, e24783. [Google Scholar] [CrossRef] [Green Version]
- Messerschmidt, D.M.; Knowles, B.B.; Solter, D. DNA methylation dynamics during epigenetic reprogramming in the germline and preimplantation embryos. Genes Dev. 2014, 28, 812–828. [Google Scholar] [CrossRef] [Green Version]
- Milagro, F.I.; Campión, J.; Cordero, P.; Goyenechea, E.; Gómez-Uriz, A.M.; Abete, I.; Zulet, M.A.; Martínez, J.A. A dual epigenomic approach for the search of obesity biomarkers: DNA methylation in relation to diet-induced weight loss. Faseb J. 2011, 25, 1378–1389. [Google Scholar] [CrossRef] [Green Version]
- Rönn, T.; Volkov, P.; Davegårdh, C.; Dayeh, T.; Hall, E.; Olsson, A.H.; Nilsson, E.; Tornberg, A.; Dekker Nitert, M.; Eriksson, K.F.; et al. A six months exercise intervention influences the genome-wide DNA methylation pattern in human adipose tissue. PLoS Genet. 2013, 9, e1003572. [Google Scholar] [CrossRef]
- Benton, M.C.; Johnstone, A.; Eccles, D.; Harmon, B.; Hayes, M.T.; Lea, R.A.; Griffiths, L.; Hoffman, E.P.; Stubbs, R.S.; Macartney-Coxson, D. An analysis of DNA methylation in human adipose tissue reveals differential modification of obesity genes before and after gastric bypass and weight loss. Genome Biol. 2015, 16, 8. [Google Scholar] [CrossRef] [Green Version]
- Barajas-Olmos, F.; Centeno-Cruz, F.; Zerrweck, C.; Imaz-Rosshandler, I.; Martínez-Hernández, A.; Cordova, E.J.; Rangel-Escareño, C.; Gálvez, F.; Castillo, A.; Maydón, H.; et al. Altered DNA methylation in liver and adipose tissues derived from individuals with obesity and type 2 diabetes. BMC Med. Genet. 2018, 19, 28. [Google Scholar] [CrossRef] [PubMed]
- Dick, K.J.; Nelson, C.P.; Tsaprouni, L.; Sandling, J.K.; Aïssi, D.; Wahl, S.; Meduri, E.; Morange, P.E.; Gagnon, F.; Grallert, H.; et al. DNA methylation and body-mass index: A genome-wide analysis. Lancet 2014, 383, 1990–1998. [Google Scholar] [CrossRef] [Green Version]
- Crujeiras, A.B.; Diaz-Lagares, A.; Moreno-Navarrete, J.M.; Sandoval, J.; Hervas, D.; Gomez, A.; Ricart, W.; Casanueva, F.F.; Esteller, M.; Fernandez-Real, J.M. Genome-wide DNA methylation pattern in visceral adipose tissue differentiates insulin-resistant from insulin-sensitive obese subjects. Transl. Res. 2016, 178, 13–24.e15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, R.F.; Thorsteinsson, E.B.; Smithson, M.; Birmingham, C.L.; Aljarallah, H.; Nolan, C. Can body temperature dysregulation explain the co-occurrence between overweight/obesity, sleep impairment, late-night eating, and a sedentary lifestyle? Eat. Weight Disord. 2017, 22, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Zimmet, P.; Alberti, K.; Stern, N.; Bilu, C.; El-Osta, A.; Einat, H.; Kronfeld-Schor, N. The Circadian Syndrome: Is the metabolic syndrome and much more! J. Intern. Med. 2019. [Google Scholar] [CrossRef]
- Nolan, C.J. Overview of the Comorbidity Between Medical Illnesses and Overweight/Obesity. In Comorbidity; Palgrave Macmillan: Cham, Switzerland, 2020; pp. 79–114. [Google Scholar] [CrossRef]
- Barton, B.B.; Zagler, A.; Engl, K.; Rihs, L.; Musil, R. Prevalence of obesity, metabolic syndrome, diabetes and risk of cardiovascular disease in a psychiatric inpatient sample: Results of the Metabolism in Psychiatry (MiP) Study. Eur. Arch. Psychiatry Clin. Neurosci. 2019. [Google Scholar] [CrossRef]
- Pi-Sunyer, X. The Look AHEAD Trial: A Review and Discussion of Its Outcomes. Curr. Nutr. Rep. 2014, 3, 387–391. [Google Scholar] [CrossRef]
- O’Donoghue, B.; Mujanovic, A.; Young, S.; Bridson, T.; Mora, L.; Bismark, M.; Cocks, J.; Siskind, D.; McGorry, P. Physical health trajectories of young people commenced on clozapine. Ir. J. Psychol. Med. 2020, 1–7. [Google Scholar] [CrossRef]
- Kim, J.A.; Kim, D.H.; Kim, S.M.; Park, Y.G.; Kim, N.H.; Baik, S.H.; Choi, K.M.; Han, K.; Yoo, H.J. Impact of the Dynamic Change of Metabolic Health Status on the Incident Type 2 Diabetes: A Nationwide Population-Based Cohort Study. Endocrinol. Metab. 2019, 34, 406–414. [Google Scholar] [CrossRef]
- Magkos, F.; Fraterrigo, G.; Yoshino, J.; Luecking, C.; Kirbach, K.; Kelly, S.C.; de Las Fuentes, L.; He, S.; Okunade, A.L.; Patterson, B.W.; et al. Effects of Moderate and Subsequent Progressive Weight Loss on Metabolic Function and Adipose Tissue Biology in Humans with Obesity. Cell Metab. 2016, 23, 591–601. [Google Scholar] [CrossRef] [Green Version]
- Liao, C.C.; Sheu, W.H.; Lin, S.Y.; Lee, W.J.; Lee, I.T. The Relationship Between Abdominal Body Composition and Metabolic Syndrome After a Weight Reduction Program in Adult Men with Obesity. Diabetes Metab. Syndr. Obes. 2020, 13, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Look Ahead Research Group. Cardiovascular effects of intensive lifestyle intervention in type 2 diabetes. N. Engl. J. Med. 2013, 369, 145–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrera, M.F.; Pantoja, J.P.; Velazquez-Fernandez, D.; Cabiedes, J.; Aguilar-Salinas, C.; Garcia-Garcia, E.; Rivas, A.; Villeda, C.; Hernandez-Ramirez, D.F.; Davila, A.; et al. Potential additional effect of omentectomy on metabolic syndrome, acute-phase reactants, and inflammatory mediators in grade III obese patients undergoing laparoscopic Roux-en-Y gastric bypass: A randomized trial. Diabetes Care 2010, 33, 1413–1418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sdralis, E.; Argentou, M.; Mead, N.; Kehagias, I.; Alexandridis, T.; Kalfarentzos, F. A prospective randomized study comparing patients with morbid obesity submitted to sleeve gastrectomy with or without omentectomy. Obes. Surg. 2013, 23, 965–971. [Google Scholar] [CrossRef] [PubMed]
- Shih, K.C.; Janckila, A.J.; Lee, W.J.; Chou, Y.C.; Huang, C.J.; Kwok, C.F.; Ho, L.T.; Chao, T.Y. Effects of bariatric weight loss surgery on glucose metabolism, inflammatory cytokines, and serum tartrate-resistant acid phosphatase 5a in obese Chinese adults. Clin. Chim. Acta. 2016, 453, 197–202. [Google Scholar] [CrossRef]
- Longo, V.D.; Panda, S. Fasting, Circadian Rhythms, and Time-Restricted Feeding in Healthy Lifespan. Cell Metab. 2016, 23, 1048–1059. [Google Scholar] [CrossRef] [Green Version]
- Smith, R.L.; Soeters, M.R.; Wust, R.C.I.; Houtkooper, R.H. Metabolic Flexibility as an Adaptation to Energy Resources and Requirements in Health and Disease. Endocr. Rev. 2018, 39, 489–517. [Google Scholar] [CrossRef] [Green Version]
- de Cabo, R.; Mattson, M.P. Effects of Intermittent Fasting on Health, Aging, and Disease. N. Engl. J. Med. 2019, 381, 2541–2551. [Google Scholar] [CrossRef]
- Malik, V.S.; Popkin, B.M.; Bray, G.A.; Despres, J.P.; Willett, W.C.; Hu, F.B. Sugar-sweetened beverages and risk of metabolic syndrome and type 2 diabetes: A meta-analysis. Diabetes Care 2010, 33, 2477–2483. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.S.; Wu, Q.J.; Xia, Y.; Zhang, J.Y.; Jiang, Y.T.; Chang, Q.; Zhao, Y.H. Carbohydrate intake and risk of metabolic syndrome: A dose-response meta-analysis of observational studies. Nutr. Metab. Cardiovasc. Dis. 2019, 29, 1288–1298. [Google Scholar] [CrossRef]
- Esposito, K.; Kastorini, C.M.; Panagiotakos, D.B.; Giugliano, D. Mediterranean diet and weight loss: Meta-analysis of randomized controlled trials. Metab. Syndr. Relat. Disord. 2011, 9, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rampelli, S.; Guenther, K.; Turroni, S.; Wolters, M.; Veidebaum, T.; Kourides, Y.; Molnár, D.; Lissner, L.; Benitez-Paez, A.; Sanz, Y.; et al. Pre-obese children’s dysbiotic gut microbiome and unhealthy diets may predict the development of obesity. Commun. Biol. 2018, 1, 222. [Google Scholar] [CrossRef] [PubMed]
- Haro, C.; Montes-Borrego, M.; Rangel-Zúñiga, O.A.; Alcalá-Díaz, J.F.; Gómez-Delgado, F.; Pérez-Martínez, P.; Delgado-Lista, J.; Quintana-Navarro, G.M.; Tinahones, F.J.; Landa, B.B.; et al. Two Healthy Diets Modulate Gut Microbial Community Improving Insulin Sensitivity in a Human Obese Population. J. Clin. Endocrin. Metab. 2016, 101, 233–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tremaroli, V.; Karlsson, F.; Werling, M.; Ståhlman, M.; Kovatcheva-Datchary, P.; Olbers, T.; Fändriks, L.; le Roux, C.W.; Nielsen, J.; Bäckhed, F. Roux-en-Y Gastric Bypass and Vertical Banded Gastroplasty Induce Long-Term Changes on the Human Gut Microbiome Contributing to Fat Mass Regulation. Cell Metab. 2015, 22, 228–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dutheil, F.; Lac, G.; Lesourd, B.; Chapier, R.; Walther, G.; Vinet, A.; Sapin, V.; Verney, J.; Ouchchane, L.; Duclos, M.; et al. Different modalities of exercise to reduce visceral fat mass and cardiovascular risk in metabolic syndrome: The RESOLVE randomized trial. Int. J. Cardiol. 2013, 168, 3634–3642. [Google Scholar] [CrossRef]
- Slentz, C.A.; Aiken, L.B.; Houmard, J.A.; Bales, C.W.; Johnson, J.L.; Tanner, C.J.; Duscha, B.D.; Kraus, W.E. Inactivity, exercise, and visceral fat. STRRIDE: A randomized, controlled study of exercise intensity and amount. J. Appl. Physiol. 2005, 99, 1613–1618. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.; Zhang, X.; Guo, J.; Roberts, C.K.; McKenzie, S.; Wu, W.C.; Liu, S.; Song, Y. Effects of Exercise Training on Cardiorespiratory Fitness and Biomarkers of Cardiometabolic Health: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. J. Am. Heart Assoc. 2015, 4. [Google Scholar] [CrossRef] [Green Version]
- Westcott, W.L. Resistance training is medicine: Effects of strength training on health. Curr Sports Med. Rep. 2012, 11, 209–216. [Google Scholar] [CrossRef]
- Steensberg, A.; Fischer, C.P.; Keller, C.; Møller, K.; Pedersen, B.K. IL-6 enhances plasma IL-1ra, IL-10, and cortisol in humans. Am. J. Physiol. Endocrinol. Metab. 2003, 285, E433–437. [Google Scholar] [CrossRef]
- Steensberg, A.; Keller, C.; Starkie, R.L.; Osada, T.; Febbraio, M.A.; Pedersen, B.K. IL-6 and TNF-alpha expression in, and release from, contracting human skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2002, 283, E1272–1278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Starkie, R.; Ostrowski, S.R.; Jauffred, S.; Febbraio, M.; Pedersen, B.K. Exercise and IL-6 infusion inhibit endotoxin-induced TNF-alpha production in humans. Faseb J. 2003, 17, 884–886. [Google Scholar] [CrossRef] [PubMed]
- Boström, P.; Wu, J.; Jedrychowski, M.P.; Korde, A.; Ye, L.; Lo, J.C.; Rasbach, K.A.; Boström, E.A.; Choi, J.H.; Long, J.Z.; et al. A PGC1-α-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature 2012, 481, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Bueno, P.G.; Bassi, D.; Contrera, D.G.; Carnielli, H.M.; Silva, R.N.; Nonaka, K.O.; Selistre-de-Araújo, H.S.; Leal, A.M. Post-exercise changes in myostatin and actRIIB expression in obese insulin-resistant rats. Mol. Cell Endocrinol. 2011, 339, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto-Mikami, E.; Sato, K.; Kurihara, T.; Hasegawa, N.; Fujie, S.; Fujita, S.; Sanada, K.; Hamaoka, T.; Tabata, I.; Iemitsu, M. Endurance training-induced increase in circulating irisin levels is associated with reduction of abdominal visceral fat in middle-aged and older adults. PLoS ONE 2015, 10, e0120354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norheim, F.; Langleite, T.M.; Hjorth, M.; Holen, T.; Kielland, A.; Stadheim, H.K.; Gulseth, H.L.; Birkeland, K.I.; Jensen, J.; Drevon, C.A. The effects of acute and chronic exercise on PGC-1α, irisin and browning of subcutaneous adipose tissue in humans. Febs J. 2014, 281, 739–749. [Google Scholar] [CrossRef] [PubMed]
- Hjorth, M.; Pourteymour, S.; Görgens, S.W.; Langleite, T.M.; Lee, S.; Holen, T.; Gulseth, H.L.; Birkeland, K.I.; Jensen, J.; Drevon, C.A.; et al. Myostatin in relation to physical activity and dysglycaemia and its effect on energy metabolism in human skeletal muscle cells. Acta Physiol. 2016, 217, 45–60. [Google Scholar] [CrossRef]
- Salas-Salvado, J.; Diaz-Lopez, A.; Ruiz-Canela, M.; Basora, J.; Fito, M.; Corella, D.; Serra-Majem, L.; Warnberg, J.; Romaguera, D.; Estruch, R.; et al. Effect of a Lifestyle Intervention Program With Energy-Restricted Mediterranean Diet and Exercise on Weight Loss and Cardiovascular Risk Factors: One-Year Results of the PREDIMED-Plus Trial. Diabetes Care 2019, 42, 777–788. [Google Scholar] [CrossRef] [Green Version]
- Mireku, M.O.; Barker, M.M.; Mutz, J.; Dumontheil, I.; Thomas, M.S.C.; Roosli, M.; Elliott, P.; Toledano, M.B. Night-time screen-based media device use and adolescents’ sleep and health-related quality of life. Environ. Int. 2019, 124, 66–78. [Google Scholar] [CrossRef]
- Dominoni, D.M.; Borniger, J.C.; Nelson, R.J. Light at night, clocks and health: From humans to wild organisms. Biol. Lett. 2016, 12, 20160015. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Zhang, L.; Zhang, Y.; Zhang, B.; He, Y.; Xie, S.; Li, M.; Miao, X.; Chan, E.Y.; Tang, J.L.; et al. Meta-analysis on night shift work and risk of metabolic syndrome. Obes. Rev. 2014, 15, 709–720. [Google Scholar] [CrossRef] [PubMed]
- Page, M.M.; Johnson, J.D. Mild Suppression of Hyperinsulinemia to Treat Obesity and Insulin Resistance. Trends Endocrinol. Metab. 2018, 29, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Corkey, B.E. Diabetes: Have we got it all wrong? Insulin hypersecretion and food additives: Cause of obesity and diabetes? Diabetes Care 2012, 35, 2432–2437. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, D.J.; Guilherme, A.; Danai, L.V.; Heyda, L.; Matevossian, A.; Cohen, J.; Nicoloro, S.M.; Straubhaar, J.; Noh, H.L.; Jung, D.; et al. A major role of insulin in promoting obesity-associated adipose tissue inflammation. Mol. Metab. 2015, 4, 507–518. [Google Scholar] [CrossRef]
- Jansen, H.J.; Stienstra, R.; van Diepen, J.A.; Hijmans, A.; van der Laak, J.A.; Vervoort, G.M.; Tack, C.J. Start of insulin therapy in patients with type 2 diabetes mellitus promotes the influx of macrophages into subcutaneous adipose tissue. Diabetologia 2013, 56, 2573–2581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Attane, C.; Peyot, M.L.; Lussier, R.; Poursharifi, P.; Zhao, S.; Zhang, D.; Morin, J.; Pineda, M.; Wang, S.; Dumortier, O.; et al. A beta cell ATGL-lipolysis/adipose tissue axis controls energy homeostasis and body weight via insulin secretion in mice. Diabetologia 2016, 59, 2654–2663. [Google Scholar] [CrossRef] [Green Version]
- Templeman, N.M.; Flibotte, S.; Chik, J.H.L.; Sinha, S.; Lim, G.E.; Foster, L.J.; Nislow, C.; Johnson, J.D. Reduced Circulating Insulin Enhances Insulin Sensitivity in Old Mice and Extends Lifespan. Cell Rep. 2017, 20, 451–463. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.Y.; Wang, J.P.; Jiang, Y.Y.; Li, H.; Hu, Y.H.; Lee, K.O.; Li, G.W. Fasting Plasma Insulin at 5 Years of Age Predicted Subsequent Weight Increase in Early Childhood over a 5-Year Period-The Da Qing Children Cohort Study. PLoS ONE 2015, 10, e0127389. [Google Scholar] [CrossRef]
- Odeleye, O.E.; de Courten, M.; Pettitt, D.J.; Ravussin, E. Fasting hyperinsulinemia is a predictor of increased body weight gain and obesity in Pima Indian children. Diabetes 1997, 46, 1341–1345. [Google Scholar] [CrossRef]
- Loves, S.; van Groningen, L.; Filius, M.; Mekking, M.; Brandon, T.; Tack, C.J.; Hermus, A.; de Boer, H. Effects of Diazoxide-Mediated Insulin Suppression on Glucose and Lipid Metabolism in Nondiabetic Obese Men. J. Clin. Endocrinol. Metab. 2018, 103, 2346–2353. [Google Scholar] [CrossRef] [Green Version]
- Velasquez-Mieyer, P.A.; Cowan, P.A.; Arheart, K.L.; Buffington, C.K.; Spencer, K.A.; Connelly, B.E.; Cowan, G.W.; Lustig, R.H. Suppression of insulin secretion is associated with weight loss and altered macronutrient intake and preference in a subset of obese adults. Int. J. Obes. Relat. Metab. Disord. 2003, 27, 219–226. [Google Scholar] [CrossRef] [Green Version]
- Takashina, C.; Tsujino, I.; Watanabe, T.; Sakaue, S.; Ikeda, D.; Yamada, A.; Sato, T.; Ohira, H.; Otsuka, Y.; Oyama-Manabe, N.; et al. Associations among the plasma amino acid profile, obesity, and glucose metabolism in Japanese adults with normal glucose tolerance. Nutr. Metab. 2016, 13, 5. [Google Scholar] [CrossRef]
- Badoud, F.; Lam, K.P.; DiBattista, A.; Perreault, M.; Zulyniak, M.A.; Cattrysse, B.; Stephenson, S.; Britz-McKibbin, P.; Mutch, D.M. Serum and adipose tissue amino acid homeostasis in the metabolically healthy obese. J. Proteome Res. 2014, 13, 3455–3466. [Google Scholar] [CrossRef] [PubMed]
- Bell, J.A.; Kivimaki, M.; Hamer, M. Metabolically healthy obesity and risk of incident type 2 diabetes: A meta-analysis of prospective cohort studies. Obes. Rev. 2014, 15, 504–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, Y.C.; Hayashi, T.; Fujimoto, W.Y.; Kahn, S.E.; Leonetti, D.L.; McNeely, M.J.; Boyko, E.J. Visceral abdominal fat accumulation predicts the conversion of metabolically healthy obese subjects to an unhealthy phenotype. Int. J. Obes. 2015, 39, 1365–1370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, Y.M.; Jung, C.H.; Cho, Y.K.; Jang, J.E.; Hwang, J.Y.; Kim, E.H.; Lee, W.J.; Park, J.Y.; Kim, H.K. Visceral adiposity index predicts the conversion of metabolically healthy obesity to an unhealthy phenotype. PLoS ONE 2017, 12, e0179635. [Google Scholar] [CrossRef] [PubMed]
- Saeed, N.; Nadeau, B.; Shannon, C.; Tincopa, M. Evaluation of Dietary Approaches for the Treatment of Non-Alcoholic Fatty Liver Disease: A Systematic Review. Nutrients 2019, 11. [Google Scholar] [CrossRef] [Green Version]
- Kite, C.; Lahart, I.M.; Afzal, I.; Broom, D.R.; Randeva, H.; Kyrou, I.; Brown, J.E. Exercise, or exercise and diet for the management of polycystic ovary syndrome: A systematic review and meta-analysis. Syst. Rev. 2019, 8, 51. [Google Scholar] [CrossRef]
- Manheimer, E.W.; van Zuuren, E.J.; Fedorowicz, Z.; Pijl, H. Paleolithic nutrition for metabolic syndrome: Systematic review and meta-analysis. Am. J. Clin. Nutr. 2015, 102, 922–932. [Google Scholar] [CrossRef] [Green Version]
- Drake, I.; Sonestedt, E.; Ericson, U.; Wallstrom, P.; Orho-Melander, M. A Western dietary pattern is prospectively associated with cardio-metabolic traits and incidence of the metabolic syndrome. Br. J. Nutr. 2018, 119, 1168–1176. [Google Scholar] [CrossRef] [Green Version]
- Chitturi, S.; Wong, V.W.; Chan, W.K.; Wong, G.L.; Wong, S.K.; Sollano, J.; Ni, Y.H.; Liu, C.J.; Lin, Y.C.; Lesmana, L.A.; et al. The Asia-Pacific Working Party on Non-alcoholic Fatty Liver Disease guidelines 2017-Part 2: Management and special groups. J. Gastroenterol. Hepatol. 2018, 33, 86–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sutton, E.F.; Beyl, R.; Early, K.S.; Cefalu, W.T.; Ravussin, E.; Peterson, C.M. Early Time-Restricted Feeding Improves Insulin Sensitivity, Blood Pressure, and Oxidative Stress Even without Weight Loss in Men with Prediabetes. Cell Metab. 2018, 27, 1212–1221.e1213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tinsley, G.M.; La Bounty, P.M. Effects of intermittent fasting on body composition and clinical health markers in humans. Nutr Rev. 2015, 73, 661–674. [Google Scholar] [CrossRef] [PubMed]
- Diabetes Prevention Program Research, G. Long-term Effects of Metformin on Diabetes Prevention: Identification of Subgroups That Benefited Most in the Diabetes Prevention Program and Diabetes Prevention Program Outcomes Study. Diabetes Care 2019, 42, 601–608. [Google Scholar] [CrossRef] [Green Version]
- Naderpoor, N.; Shorakae, S.; de Courten, B.; Misso, M.L.; Moran, L.J.; Teede, H.J. Metformin and lifestyle modification in polycystic ovary syndrome: Systematic review and meta-analysis. Hum. Reprod. Update 2016, 22, 408–409. [Google Scholar] [CrossRef] [Green Version]
- Cheng, H.S.; Tan, W.R.; Low, Z.S.; Marvalim, C.; Lee, J.Y.H.; Tan, N.S. Exploration and Development of PPAR Modulators in Health and Disease: An Update of Clinical Evidence. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef] [Green Version]
- Tan, C.K.; Zhuang, Y.; Wahli, W. Synthetic and natural Peroxisome Proliferator-Activated Receptor (PPAR) agonists as candidates for the therapy of the metabolic syndrome. Expert Opin. Ther. Targets. 2017, 21, 333–348. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Inzucchi, S.; Abdul-Ghani, M.; Nissen, S.E. Pioglitazone: The forgotten, cost-effective cardioprotective drug for type 2 diabetes. Diab. Vasc. Dis. Res. 2019, 16, 133–143. [Google Scholar] [CrossRef] [Green Version]
- Tolman, K.G. The safety of thiazolidinediones. Expert Opin. Drug. Saf. 2011, 10, 419–428. [Google Scholar] [CrossRef]
- Fruchart, J.C.; Santos, R.D.; Aguilar-Salinas, C.; Aikawa, M.; Al Rasadi, K.; Amarenco, P.; Barter, P.J.; Ceska, R.; Corsini, A.; Despres, J.P.; et al. The selective peroxisome proliferator-activated receptor alpha modulator (SPPARMalpha) paradigm: Conceptual framework and therapeutic potential: A consensus statement from the International Atherosclerosis Society (IAS) and the Residual Risk Reduction Initiative (R3i) Foundation. Cardiovasc. Diabetol. 2019, 18, 71. [Google Scholar] [CrossRef]
- Sasaki, Y.; Asahiyama, M.; Tanaka, T.; Yamamoto, S.; Murakami, K.; Kamiya, W.; Matsumura, Y.; Osawa, T.; Anai, M.; Fruchart, J.C.; et al. Pemafibrate, a selective PPARalpha modulator, prevents non-alcoholic steatohepatitis development without reducing the hepatic triglyceride content. Sci. Rep. 2020, 10, 7818. [Google Scholar] [CrossRef] [PubMed]
- Kaul, U.; Parmar, D.; Manjunath, K.; Shah, M.; Parmar, K.; Patil, K.P.; Jaiswal, A. New dual peroxisome proliferator activated receptor agonist-Saroglitazar in diabetic dyslipidemia and non-alcoholic fatty liver disease: Integrated analysis of the real world evidence. Cardiovasc. Diabetol. 2019, 18, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhosle, D.; Bhosle, V.; Bobde, J.; Bhagat, A.; Shaikh, H.; Kadam, R. Study of Saroglitazar in Treatment Of Pre-diabetes with Dyslipidemia: STOP-D. J. Assoc. Physicians India 2018, 66, 14–17. [Google Scholar] [PubMed]
- Davies, M.J.; D’Alessio, D.A.; Fradkin, J.; Kernan, W.N.; Mathieu, C.; Mingrone, G.; Rossing, P.; Tsapas, A.; Wexler, D.J.; Buse, J.B. Management of hyperglycaemia in type 2 diabetes, 2018. A consensus report by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetologia 2018, 61, 2461–2498. [Google Scholar] [CrossRef] [Green Version]
- Burcelin, R.; Gourdy, P. Harnessing glucagon-like peptide-1 receptor agonists for the pharmacological treatment of overweight and obesity. Obes. Rev. 2017, 18, 86–98. [Google Scholar] [CrossRef]
- Nylander, M.; Frossing, S.; Clausen, H.V.; Kistorp, C.; Faber, J.; Skouby, S.O. Effects of liraglutide on ovarian dysfunction in polycystic ovary syndrome: A randomized clinical trial. Reprod. Biomed. Online 2017, 35, 121–127. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, M.J.; Gaunt, P.; Aithal, G.P.; Barton, D.; Hull, D.; Parker, R.; Hazlehurst, J.M.; Guo, K.; team, L.t.; Abouda, G.; et al. Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): A multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet 2016, 387, 679–690. [Google Scholar] [CrossRef] [Green Version]
- Kelly, A.S.; Auerbach, P.; Barrientos-Perez, M.; Gies, I.; Hale, P.M.; Marcus, C.; Mastrandrea, L.D.; Prabhu, N.; Arslanian, S.; Investigators, N.N.T. A Randomized, Controlled Trial of Liraglutide for Adolescents with Obesity. N. Engl. J. Med. 2020, 382, 2117–2128. [Google Scholar] [CrossRef]
- Sanchez-Garrido, M.A.; Brandt, S.J.; Clemmensen, C.; Muller, T.D.; DiMarchi, R.D.; Tschop, M.H. GLP-1/glucagon receptor co-agonism for treatment of obesity. Diabetologia 2017, 60, 1851–1861. [Google Scholar] [CrossRef]
- Frias, J.P.; Nauck, M.A.; Van, J.; Kutner, M.E.; Cui, X.; Benson, C.; Urva, S.; Gimeno, R.E.; Milicevic, Z.; Robins, D.; et al. Efficacy and safety of LY3298176, a novel dual GIP and GLP-1 receptor agonist, in patients with type 2 diabetes: A randomised, placebo-controlled and active comparator-controlled phase 2 trial. Lancet 2018, 392, 2180–2193. [Google Scholar] [CrossRef]
- Hartman, M.L.; Sanyal, A.J.; Loomba, R.; Wilson, J.M.; Nikooienejad, A.; Bray, R.; Karanikas, C.A.; Duffin, K.L.; Robins, D.A.; Haupt, A. Effects of Novel Dual GIP and GLP-1 Receptor Agonist Tirzepatide on Biomarkers of Nonalcoholic Steatohepatitis in Patients With Type 2 Diabetes. Diabetes Care 2020, 43, 1352–1355. [Google Scholar] [CrossRef] [PubMed]
- Rebello, C.J.; Greenway, F.L. Obesity medications in development. Expert Opin. Investig. Drugs 2020, 29, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Athyros, V.G.; Tziomalos, K.; Karagiannis, A.; Mikhailidis, D.P. Cardiovascular benefits of bariatric surgery in morbidly obese patients. Obes. Rev. 2011, 12, 515–524. [Google Scholar] [CrossRef] [PubMed]
- Reed, M.A.; Pories, W.J.; Chapman, W.; Pender, J.; Bowden, R.; Barakat, H.; Gavin, T.P.; Green, T.; Tapscott, E.; Zheng, D.; et al. Roux-en-Y gastric bypass corrects hyperinsulinemia implications for the remission of type 2 diabetes. J. Clin. Endocrinol. Metab. 2011, 96, 2525–2531. [Google Scholar] [CrossRef] [Green Version]
- Alemzadeh, R.; Langley, G.; Upchurch, L.; Smith, P.; Slonim, A.E. Beneficial effect of diazoxide in obese hyperinsulinemic adults. J. Clin. Endocrinol. Metab. 1998, 83, 1911–1915. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dagpo, T.D.; Nolan, C.J.; Delghingaro-Augusto, V. Exploring Therapeutic Targets to Reverse or Prevent the Transition from Metabolically Healthy to Unhealthy Obesity. Cells 2020, 9, 1596. https://doi.org/10.3390/cells9071596
Dagpo TD, Nolan CJ, Delghingaro-Augusto V. Exploring Therapeutic Targets to Reverse or Prevent the Transition from Metabolically Healthy to Unhealthy Obesity. Cells. 2020; 9(7):1596. https://doi.org/10.3390/cells9071596
Chicago/Turabian StyleDagpo, Tenzin D., Christopher J. Nolan, and Viviane Delghingaro-Augusto. 2020. "Exploring Therapeutic Targets to Reverse or Prevent the Transition from Metabolically Healthy to Unhealthy Obesity" Cells 9, no. 7: 1596. https://doi.org/10.3390/cells9071596
APA StyleDagpo, T. D., Nolan, C. J., & Delghingaro-Augusto, V. (2020). Exploring Therapeutic Targets to Reverse or Prevent the Transition from Metabolically Healthy to Unhealthy Obesity. Cells, 9(7), 1596. https://doi.org/10.3390/cells9071596