PCAF Involvement in Lamin A/C-HDAC2 Interplay during the Early Phase of Muscle Differentiation


 ,
,  and
and 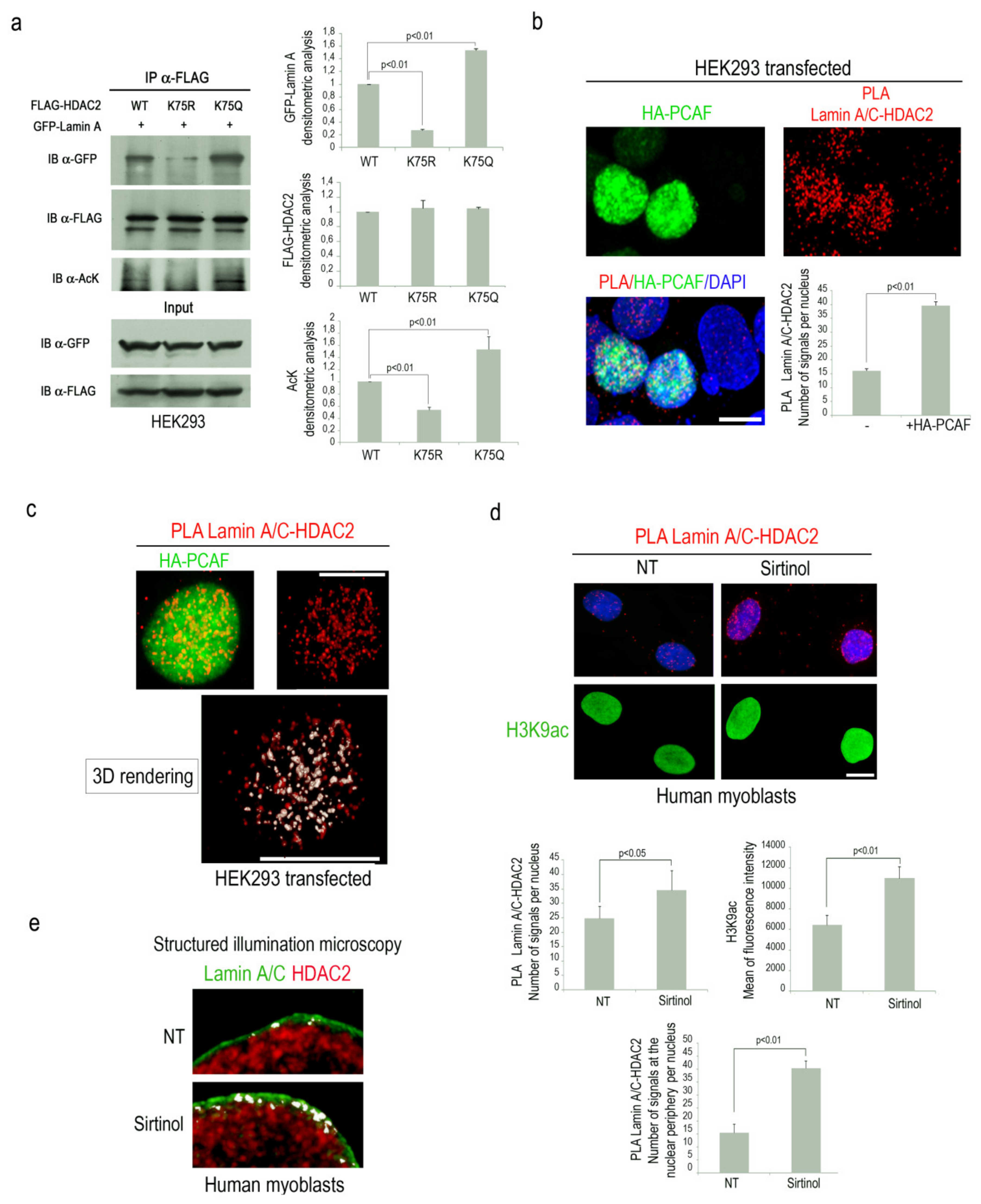{kind=link}
{kind=link}
{kind=link}
{kind=link}
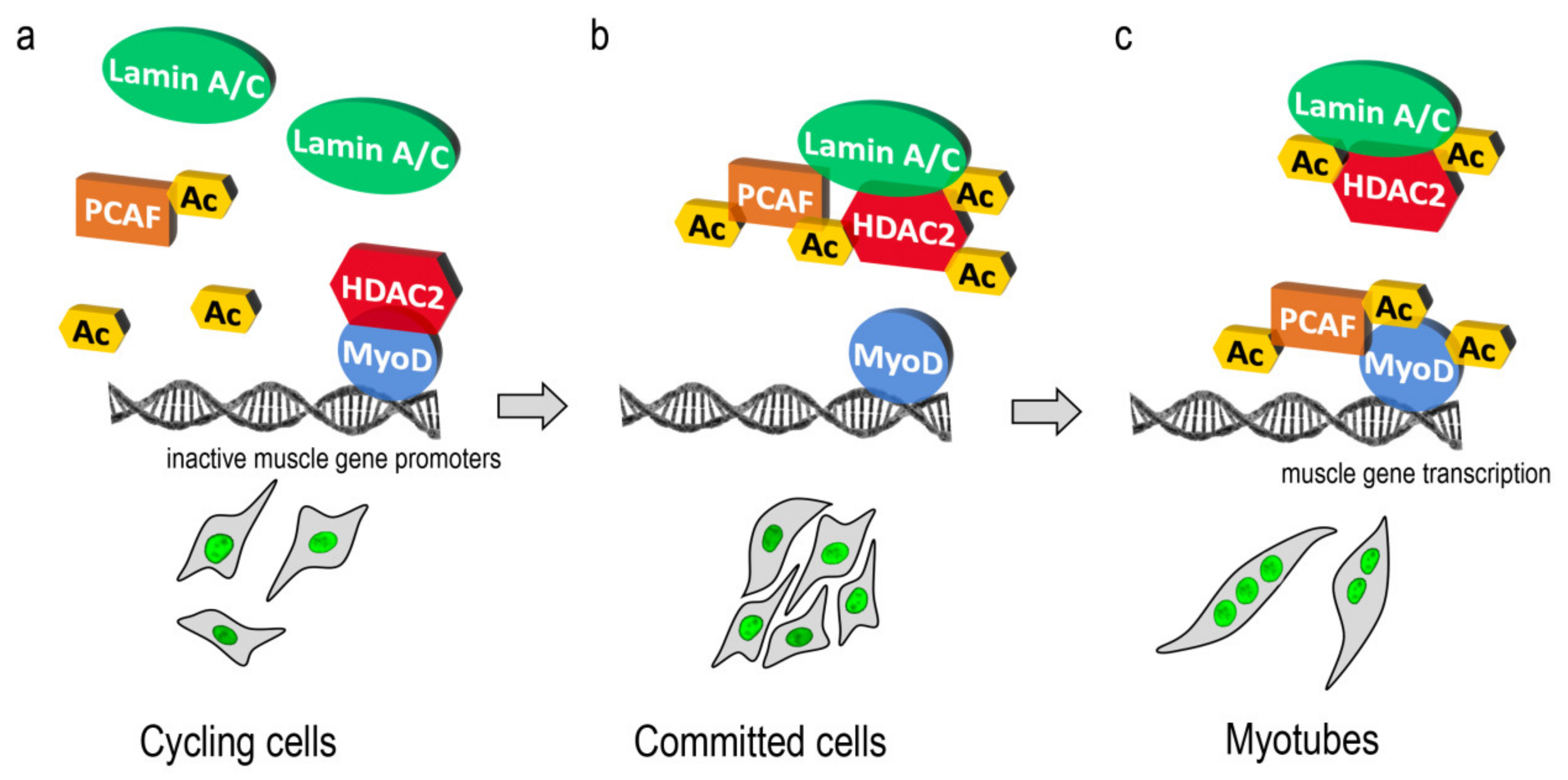{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture, Differentiation and Transfection
- mHDAC2-K75R-F: 5’-AGCGATGAGTATATCAGGTTTCTACGATCAATA-3’
- mHDAC2 K75R-R: 5’-TATTGATCGTAGAAACCTGATATACTCATCGCT-3’
- mHDCA2-K75Q-F: 5’-AGCGATGAGTATATCCAGTTTCTACGATCAATA-3’
- mHDCA2-K75Q-R: 5’-TATTGATCGTAGAAACTGGATATACTCATCGCT-3’
2.2. Antibodies and Drugs
2.3. In Situ Proximity Ligation Assay
2.4. Co-Immunoprecipitation Experiments
2.5. Immunofluorescence Analysis
2.6. Imaging
3. Results
3.1. PCAF Promotes HDAC2-Lamin A/C Interaction
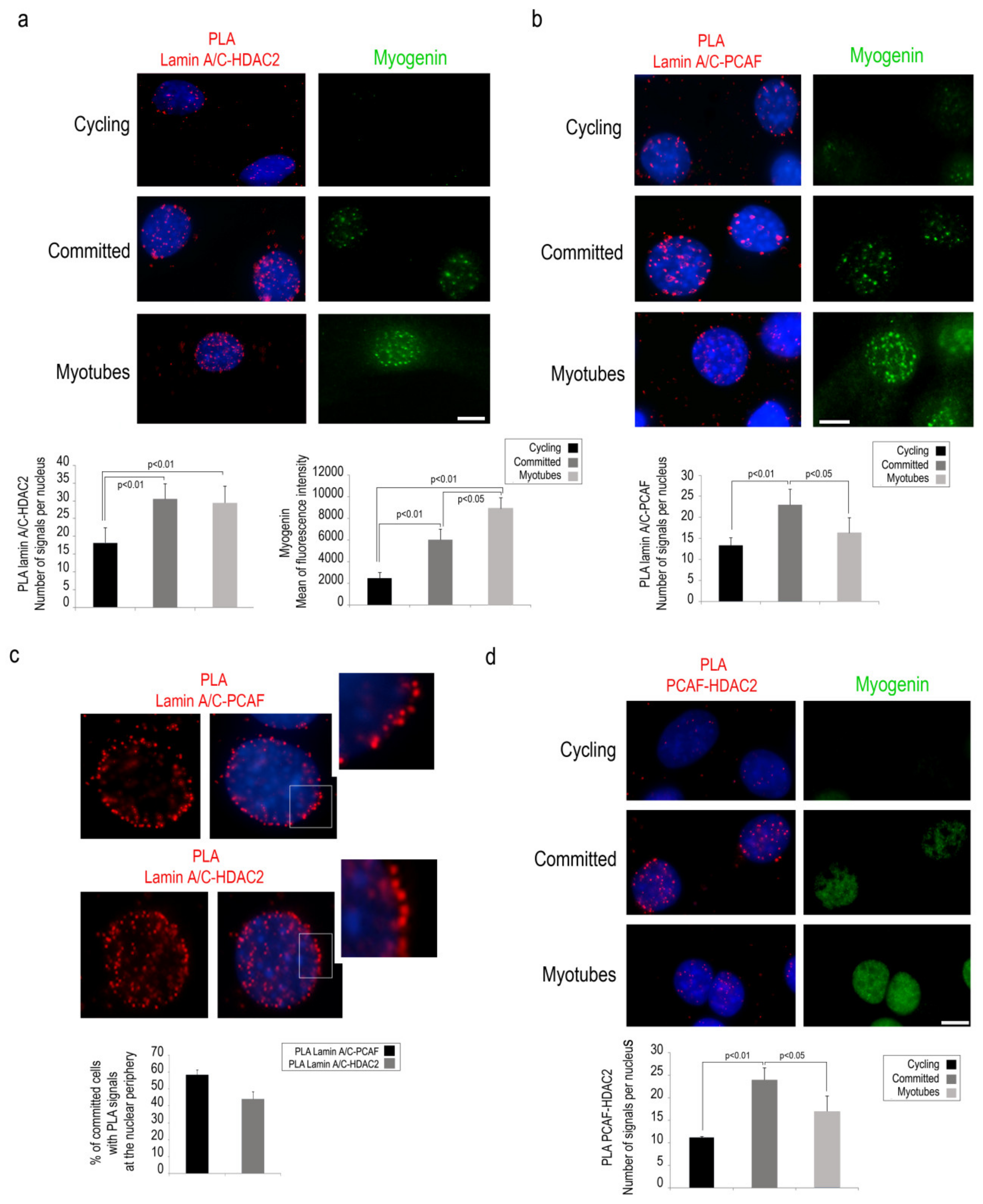
3.2. PCAF-Lamin A/C Interaction During the Early Phase of Muscle Differentiation
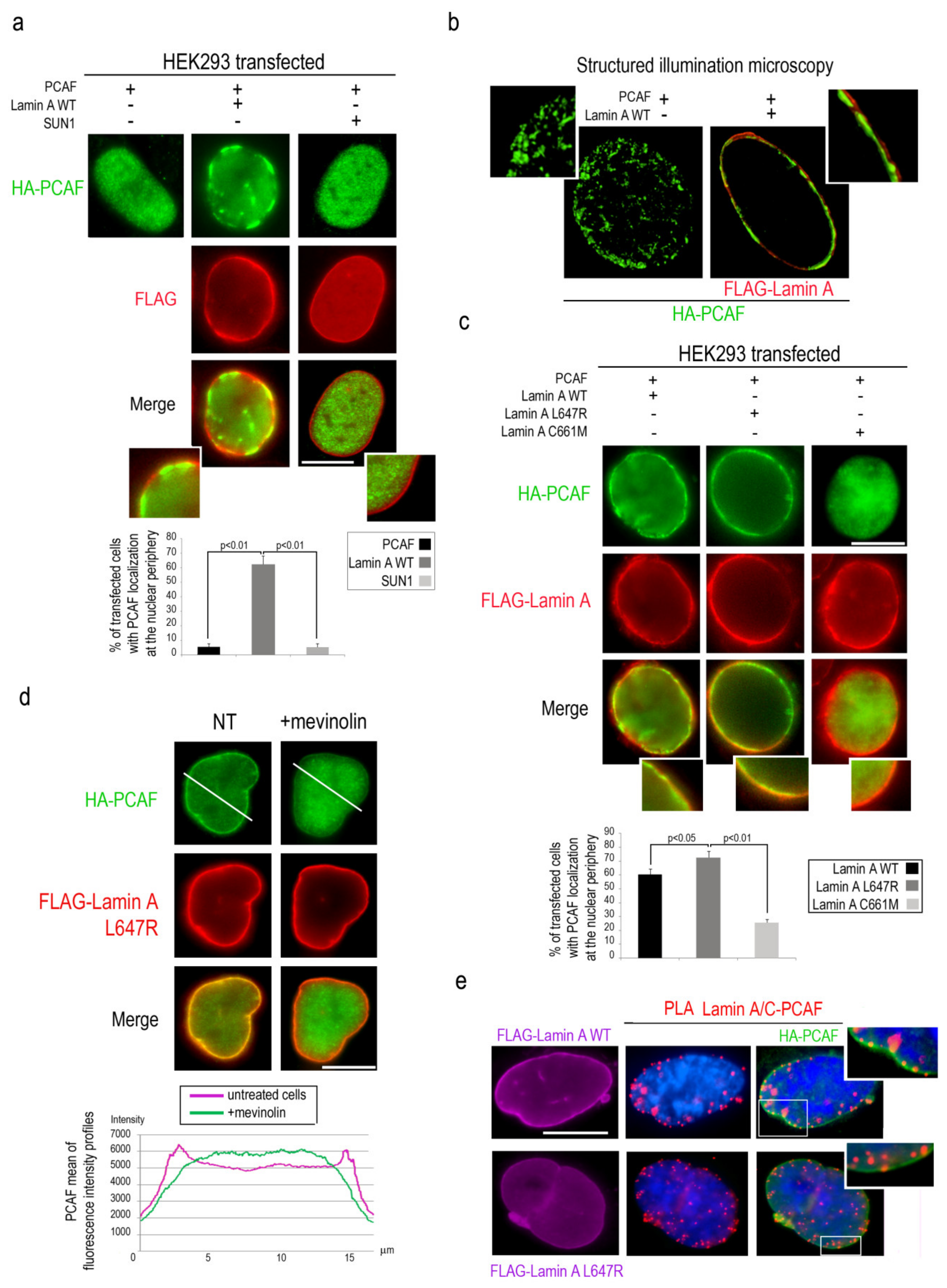
3.3. Lamin A Recruits PCAF to the Nuclear Lamina
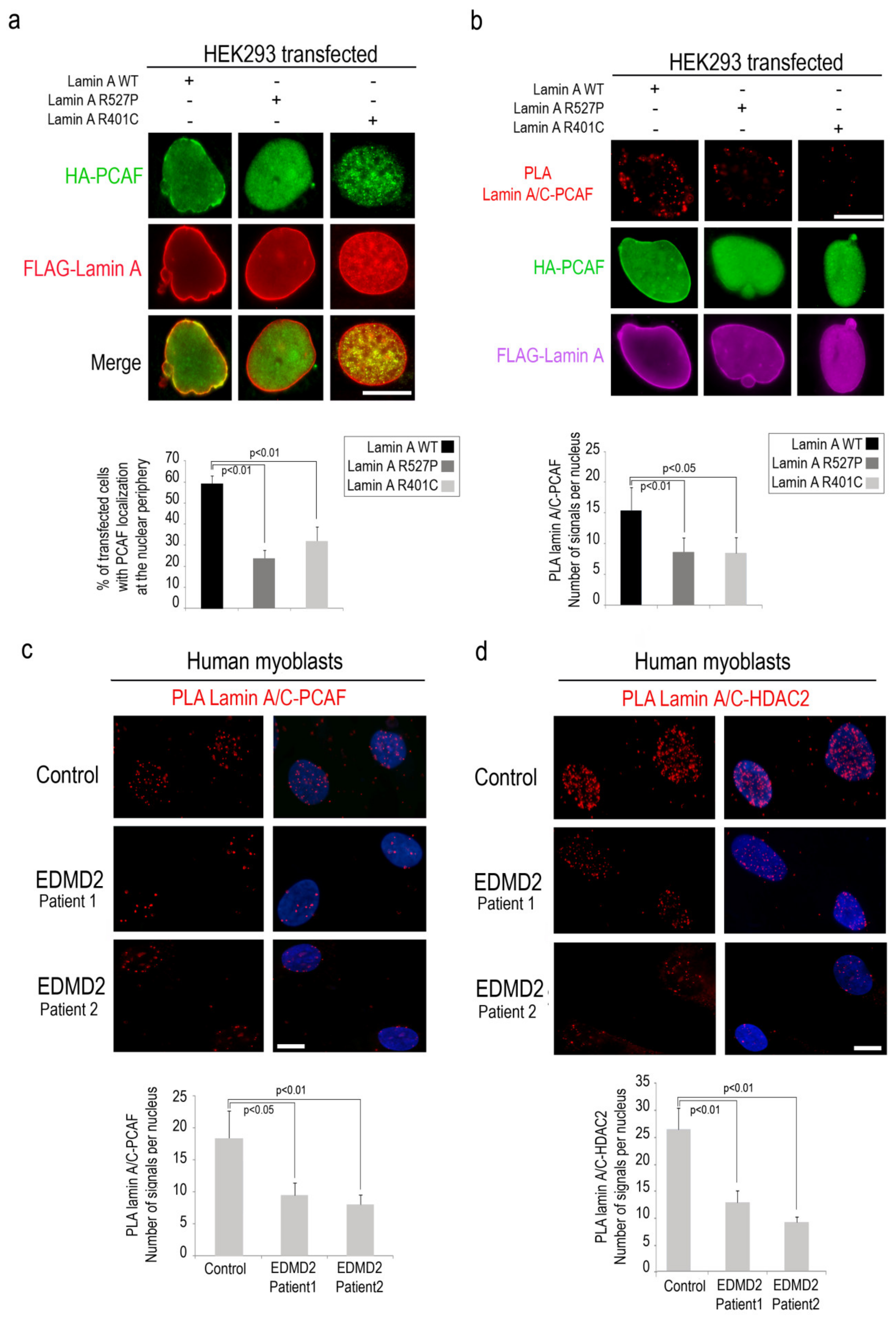
3.4. PCAF-Lamin A/C Interaction is Altered by LMNA Mutations Causing EDMD Phenotype
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Moresi, V.; Marroncelli, N.; Coletti, D.; Adamo, S. Regulation of skeletal muscle development and homeostasis by gene imprinting, histone acetylation and microRNA. Biochim. Biophys. Acta BBA Gene Regul. Mech. 2015, 1894, 309–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puri, P.L.; Iezzi, S.; Stiegler, P.; Chen, T.-T.; Schiltz, R.L.; Muscat, G.E.O.; Giordano, A.; Kedes, L.; Wang, J.Y.J.; Sartorelli, V. Class I Histone Deacetylases Sequentially Interact with MyoD and pRb during Skeletal Myogenesis. Mol. Cell 2001, 8, 885–897. [Google Scholar] [CrossRef]
- Seto, E.; Yoshida, M. Erasers of Histone Acetylation: The Histone Deacetylase Enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef] [Green Version]
- Eom, G.H.; Nam, Y.S.; Oh, J.G.; Choe, N.; Min, H.-K.; Yoo, E.-K.; Kang, G.; Nguyen, V.H.; Min, J.-J.; Kim, J.-K.; et al. Regulation of Acetylation of Histone Deacetylase 2 by p300/CBP-Associated Factor/Histone Deacetylase 5 in the Development of Cardiac Hypertrophy. Circ. Res. 2014, 114, 1133–1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, O.H.; Mallappa, C.; Hernández-Hernández, J.M.; Rivera-Pérez, J.A.; Imbalzano, A.N. Contrasting roles for MyoD in organizing myogenic promoter structures during embryonic skeletal muscle development: Contrasting Functions for Myod. Dev. Dyn. 2015, 244, 43–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sartorelli, V.; Puri, P.L.; Hamamori, Y.; Ogryzko, V.; Chung, G.; Nakatani, Y.; Wang, J.Y.J.; Kedes, L. Acetylation of MyoD Directed by PCAF Is Necessary for the Execution of the Muscle Program. Mol. Cell 1999, 4, 725–734. [Google Scholar] [CrossRef]
- Soleimani, V.D.; Yin, H.; Jahani-Asl, A.; Ming, H.; Kockx, C.E.M.; van Ijcken, W.F.J.; Grosveld, F.; Rudnicki, M.A. Snail Regulates MyoD Binding-Site Occupancy to Direct Enhancer Switching and Differentiation-Specific Transcription in Myogenesis. Mol. Cell 2012, 47, 457–468. [Google Scholar] [CrossRef] [Green Version]
- Ohkawa, Y.; Marfella, C.G.A.; Imbalzano, A.N. Skeletal muscle specification by myogenin and Mef2D via the SWI/SNF ATPase Brg1. EMBO J. 2006, 25, 490–501. [Google Scholar] [CrossRef] [Green Version]
- Dilworth, F.J.; Seaver, K.J.; Fishburn, A.L.; Htet, S.L.; Tapscott, S.J. In vitro transcription system delineates the distinct roles of the coactivators pCAF and p300 during MyoD/E47-dependent transactivation. Proc. Natl. Acad. Sci. USA 2004, 101, 11593–11598. [Google Scholar] [CrossRef] [Green Version]
- Davies, B.S.J.; Fong, L.G.; Yang, S.H.; Coffinier, C.; Young, S.G. The Posttranslational Processing of Prelamin A and Disease. Annu. Rev. Genom. Hum. Genet. 2009, 10, 153–174. [Google Scholar] [CrossRef] [Green Version]
- Mattioli, E.; Columbaro, M.; Capanni, C.; Santi, S.; Maraldi, N.M.; D’Apice, M.R.; Novelli, G.; Riccio, M.; Squarzoni, S.; Foisner, R.; et al. Drugs affecting prelamin A processing: Effects on heterochromatin organization. Exp. Cell Res. 2008, 314, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Dubinska-Magiera, M.; Zaremba-Czogalla, M.; Rzepecki, R. Muscle development, regeneration and laminopathies: How lamins or lamina-associated proteins can contribute to muscle development, regeneration and disease. Cell. Mol. Life Sci. 2013, 70, 2713–2741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frock, R.L. Lamin A/C and emerin are critical for skeletal muscle satellite cell differentiation. Genes Dev. 2006, 20, 486–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gómez-Domínguez, D.; Epifano, C.; de Miguel, F.; Castaño, A.G.; Vilaplana-Martí, B.; Martín, A.; Amarilla-Quintana, S.; Bertrand, A.T.; Bonne, G.; Ramón-Azcón, J.; et al. Consequences of Lmna Exon 4 Mutations in Myoblast Function. Cells 2020, 9, 1286. [Google Scholar] [CrossRef]
- Athar, F.; Parnaik, V.K. Association of lamin A/C with muscle gene-specific promoters in myoblasts. Biochem. Biophys. Rep. 2015, 4, 76–82. [Google Scholar] [CrossRef] [Green Version]
- Capanni, C.; Del Coco, R.; Squarzoni, S.; Columbaro, M.; Mattioli, E.; Camozzi, D.; Rocchi, A.; Scotlandi, K.; Maraldi, N.; Foisner, R. Prelamin A is involved in early steps of muscle differentiation. Exp. Cell Res. 2008, 314, 3628–3637. [Google Scholar] [CrossRef]
- Mattioli, E.; Columbaro, M.; Capanni, C.; Maraldi, N.M.; Cenni, V.; Scotlandi, K.; Marino, M.T.; Merlini, L.; Squarzoni, S.; Lattanzi, G. Prelamin A-mediated recruitment of SUN1 to the nuclear envelope directs nuclear positioning in human muscle. Cell Death Differ. 2011, 18, 1305–1315. [Google Scholar] [CrossRef]
- Angori, S.; Capanni, C.; Faulkner, G.; Bean, C.; Boriani, G.; Lattanzi, G.; Cenni, V. Emery-Dreifuss Muscular Dystrophy-Associated Mutant Forms of Lamin A Recruit the Stress Responsive Protein Ankrd2 into the Nucleus, Affecting the Cellular Response to Oxidative Stress. Cell Physiol. Biochem. 2017, 42, 169–184. [Google Scholar] [CrossRef]
- Cenni, V.; Kojic, S.; Capanni, C.; Faulkner, G.; Lattanzi, G. Ankrd2 in Mechanotransduction and Oxidative Stress Response in Skeletal Muscle: New Cues for the Pathogenesis of Muscular Laminopathies. Oxidative Med. Cell. Longev. 2019, 2019, 1–15. [Google Scholar] [CrossRef]
- Perovanovic, J.; Dell’Orso, S.; Gnochi, V.F.; Jaiswal, J.K.; Sartorelli, V.; Vigouroux, C.; Mamchaoui, K.; Mouly, V.; Bonne, G.; Hoffman, E.P. Laminopathies disrupt epigenomic developmental programs and cell fate. Sci. Transl. Med. 2016, 8, 335ra58. [Google Scholar] [CrossRef] [Green Version]
- Håkelien, A.-M.; Delbarre, E.; Gaustad, K.G.; Buendia, B.; Collas, P. Expression of the myodystrophic R453W mutation of lamin A in C2C12 myoblasts causes promoter-specific and global epigenetic defects. Exp. Cell Res. 2008, 314, 1869–1880. [Google Scholar] [CrossRef] [PubMed]
- Melcon, G.; Kozlov, S.; Cutler, D.A.; Sullivan, T.; Hernandez, L.; Zhao, P.; Mitchell, S.; Nader, G.; Bakay, M.; Rottman, J.N.; et al. Loss of emerin at the nuclear envelope disrupts the Rb1/E2F and MyoD pathways during muscle regeneration. Hum. Mol. Genet. 2006, 15, 637–651. [Google Scholar] [CrossRef] [Green Version]
- Bakay, M.; Wang, Z.; Melcon, G.; Schiltz, L.; Xuan, J.; Zhao, P.; Sartorelli, V.; Seo, J.; Pegoraro, E.; Angelini, C.; et al. Nuclear envelope dystrophies show a transcriptional fingerprint suggesting disruption of Rb–MyoD pathways in muscle regeneration. Brain 2006, 129, 996–1013. [Google Scholar] [CrossRef] [PubMed]
- Mattioli, E.; Andrenacci, D.; Garofalo, C.; Prencipe, S.; Scotlandi, K.; Remondini, D.; Gentilini, D.; Di Blasio, A.M.; Valente, S.; Scarano, E.; et al. Altered modulation of lamin A/C-HDAC2 interaction and p21 expression during oxidative stress response in HGPS. Aging Cell 2018, 17, e12824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattioli, E.; Andrenacci, D.; Mai, A.; Valente, S.; Robijns, J.; De Vos, W.H.; Capanni, C.; Lattanzi, G. Statins and Histone Deacetylase Inhibitors Affect Lamin A/C–Histone Deacetylase 2 Interaction in Human Cells. Front. Cell Dev. Biol. 2019, 7, 6. [Google Scholar] [CrossRef]
- Bianchi, A.; Mozzetta, C.; Pegoli, G.; Lucini, F.; Valsoni, S.; Rosti, V.; Petrini, C.; Cortesi, A.; Gregoretti, F.; Antonelli, L.; et al. Dysfunctional polycomb transcriptional repression contributes to lamin A/C–dependent muscular dystrophy. J. Clin. Investig. 2020, 130, 2408–2421. [Google Scholar] [CrossRef] [Green Version]
- Meinke, P.; Kerr, A.R.W.; Czapiewski, R.; de las Heras, J.I.; Dixon, C.R.; Harris, E.; Kölbel, H.; Muntoni, F.; Schara, U.; Straub, V.; et al. A multistage sequencing strategy pinpoints novel candidate alleles for Emery-Dreifuss muscular dystrophy and supports gene misregulation as its pathomechanism. eBioMedicine 2020, 51, 102587. [Google Scholar] [CrossRef] [Green Version]
- Cesarini, E.; Mozzetta, C.; Marullo, F.; Gregoretti, F.; Gargiulo, A.; Columbaro, M.; Cortesi, A.; Antonelli, L.; Di Pelino, S.; Squarzoni, S.; et al. Lamin A/C sustains PcG protein architecture, maintaining transcriptional repression at target genes. J. Cell Biol. 2015, 211, 533–551. [Google Scholar] [CrossRef] [Green Version]
- Bossone, K.A.; Ellis, J.A.; Holaska, J.M. Histone acetyltransferase inhibition rescues differentiation of emerin-deficient myogenic progenitors. Muscle Nerve 2020, 62, 128–136. [Google Scholar] [CrossRef]
- Peng, S.; Zhao, S.; Yan, F.; Cheng, J.; Huang, L.; Chen, H.; Liu, Q.; Ji, X.; Yuan, Z. HDAC2 Selectively Regulates FOXO3a-Mediated Gene Transcription during Oxidative Stress-Induced Neuronal Cell Death. J. Neurosci. 2015, 35, 1250–1259. [Google Scholar] [CrossRef] [Green Version]
- Fulco, M.; Schiltz, R.L.; Iezzi, S.; King, M.T.; Zhao, P.; Kashiwaya, Y.; Hoffman, E.; Veech, R.L.; Sartorelli, V. Sir2 Regulates Skeletal Muscle Differentiation as a Potential Sensor of the Redox State. Mol. Cell 2003, 12, 51–62. [Google Scholar] [CrossRef]
- Riccio, M.; Dembic, M.; Cinti, C.; Santi, S. Multifluorescence Labeling and Colocalization Analyses. In Cell Cycle Control and Dysregulation Protocols; Humana Press: Totowa, NJ, USA, 2004; Volume 285, pp. 171–178. ISBN 978-1-59259-822-9. [Google Scholar]
- Vignoli, B.; Battistini, G.; Melani, R.; Blum, R.; Santi, S.; Berardi, N.; Canossa, M. Peri-Synaptic Glia Recycles Brain-Derived Neurotrophic Factor for LTP Stabilization and Memory Retention. Neuron 2016, 92, 873–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pediconi, N.; Guerrieri, F.; Vossio, S.; Bruno, T.; Belloni, L.; Schinzari, V.; Scisciani, C.; Fanciulli, M.; Levrero, M. hSirT1-Dependent Regulation of the PCAF-E2F1-p73 Apoptotic Pathway in Response to DNA Damage. MCB 2009, 29, 1989–1998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, P.; Liu, Y.; Jin, C.; Zhang, M.; Lv, L.; Zhang, X.; Liu, H.; Zhou, Y. Histone H3K9 Acetyltransferase PCAF Is Essential for Osteogenic Differentiation Through Bone Morphogenetic Protein Signaling and May Be Involved in Osteoporosis: PCAF Promotes Osteogenic Differentiation. Stem Cells 2016, 34, 2332–2341. [Google Scholar] [CrossRef] [PubMed]
- Yamagoe, S.; Kanno, T.; Kanno, Y.; Sasaki, S.; Siegel, R.M.; Lenardo, M.J.; Humphrey, G.; Wang, Y.; Nakatani, Y.; Howard, B.H.; et al. Interaction of Histone Acetylases and Deacetylases In Vivo. MCB 2003, 23, 1025–1033. [Google Scholar] [CrossRef] [Green Version]
- Savoia, M.; Cencioni, C.; Mori, M.; Atlante, S.; Zaccagnini, G.; Devanna, P.; Di Marcotullio, L.; Botta, B.; Martelli, F.; Zeiher, A.M.; et al. P300/CBP-associated factor regulates transcription and function of isocitrate dehydrogenase 2 during muscle differentiation. FASEB J. 2019, 33, 4107–4123. [Google Scholar] [CrossRef]
- Puri, P.L.; Sartorelli, V.; Yang, X.-J.; Hamamori, Y.; Ogryzko, V.V.; Howard, B.H.; Kedes, L.; Wang, J.Y.J.; Graessmann, A.; Nakatani, Y.; et al. Differential Roles of p300 and PCAF Acetyltransferases in Muscle Differentiation. Mol. Cell 1997, 1, 35–45. [Google Scholar] [CrossRef]
- Iezzi, S.; Cossu, G.; Nervi, C.; Sartorelli, V.; Puri, P.L. Stage-specific modulation of skeletal myogenesis by inhibitors of nuclear deacetylases. Proc. Natl. Acad. Sci. USA 2002, 99, 7757–7762. [Google Scholar] [CrossRef] [Green Version]
- Sincennes, M.-C.; Brun, C.E.; Rudnicki, M.A. Concise Review: Epigenetic Regulation of Myogenesis in Health and Disease: Epigenetic Regulation of Myogenesis. Stem Cells Transl. Med. 2016, 5, 282–290. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santi, S.; Cenni, V.; Capanni, C.; Lattanzi, G.; Mattioli, E. PCAF Involvement in Lamin A/C-HDAC2 Interplay during the Early Phase of Muscle Differentiation. Cells 2020, 9, 1735. https://doi.org/10.3390/cells9071735
Santi S, Cenni V, Capanni C, Lattanzi G, Mattioli E. PCAF Involvement in Lamin A/C-HDAC2 Interplay during the Early Phase of Muscle Differentiation. Cells. 2020; 9(7):1735. https://doi.org/10.3390/cells9071735
Chicago/Turabian StyleSanti, Spartaco, Vittoria Cenni, Cristina Capanni, Giovanna Lattanzi, and Elisabetta Mattioli. 2020. "PCAF Involvement in Lamin A/C-HDAC2 Interplay during the Early Phase of Muscle Differentiation" Cells 9, no. 7: 1735. https://doi.org/10.3390/cells9071735
APA StyleSanti, S., Cenni, V., Capanni, C., Lattanzi, G., & Mattioli, E. (2020). PCAF Involvement in Lamin A/C-HDAC2 Interplay during the Early Phase of Muscle Differentiation. Cells, 9(7), 1735. https://doi.org/10.3390/cells9071735