Lysophosphatidic Acid Receptor 1- and 3-Mediated Hyperalgesia and Hypoalgesia in Diabetic Neuropathic Pain Models in Mice



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
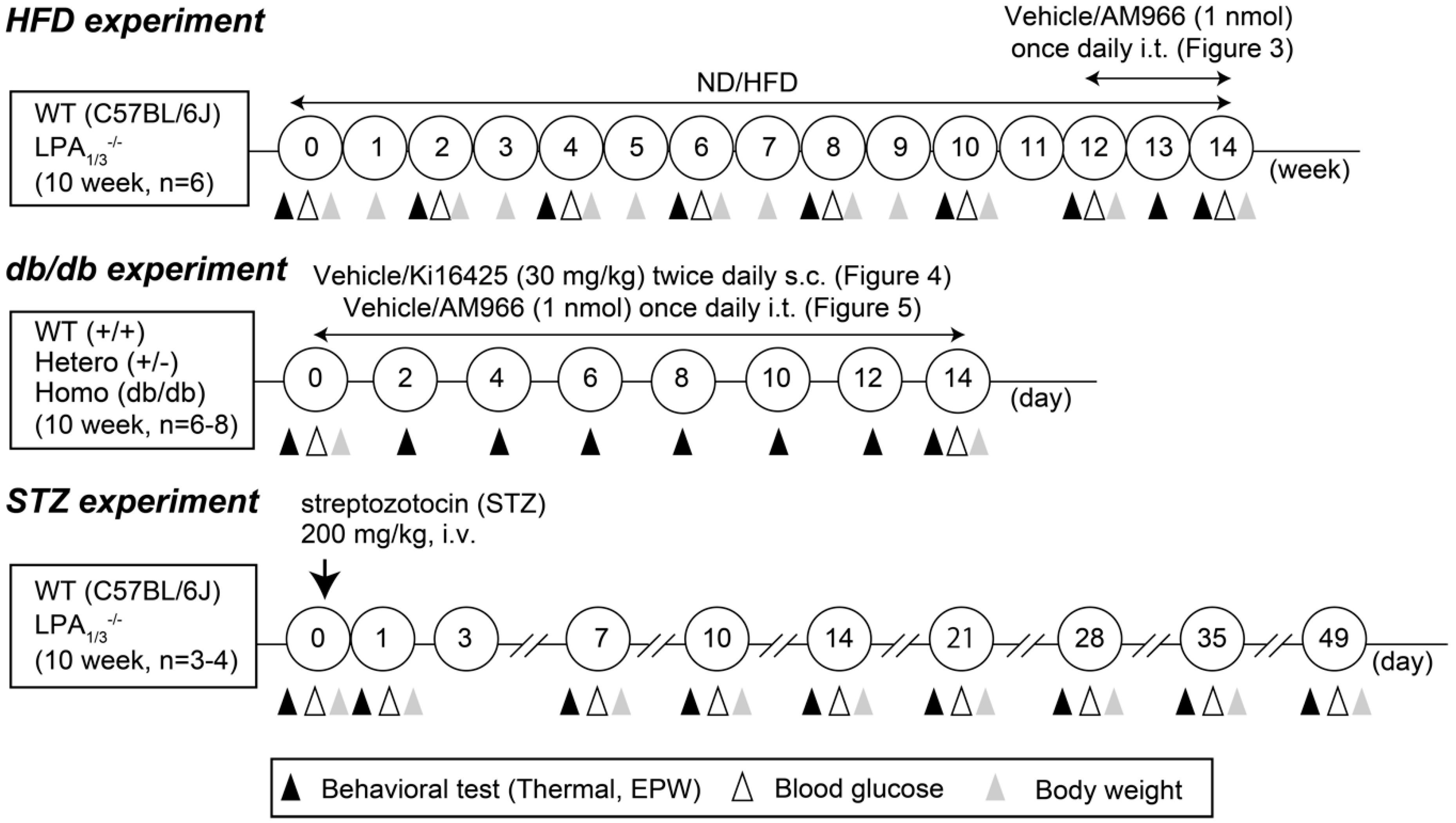
2. Materials and Methods
2.1. Drugs
2.2. Animals
2.3. Thermal Paw Withdrawal Test
2.4. Electrical Stimulation-Induced Paw Withdrawal (EPW) Test
2.5. Mechanical Paw Pressure Test and von Frey Filament Test
2.6. Blood Glucose Measurement
2.7. Statistics
3. Results
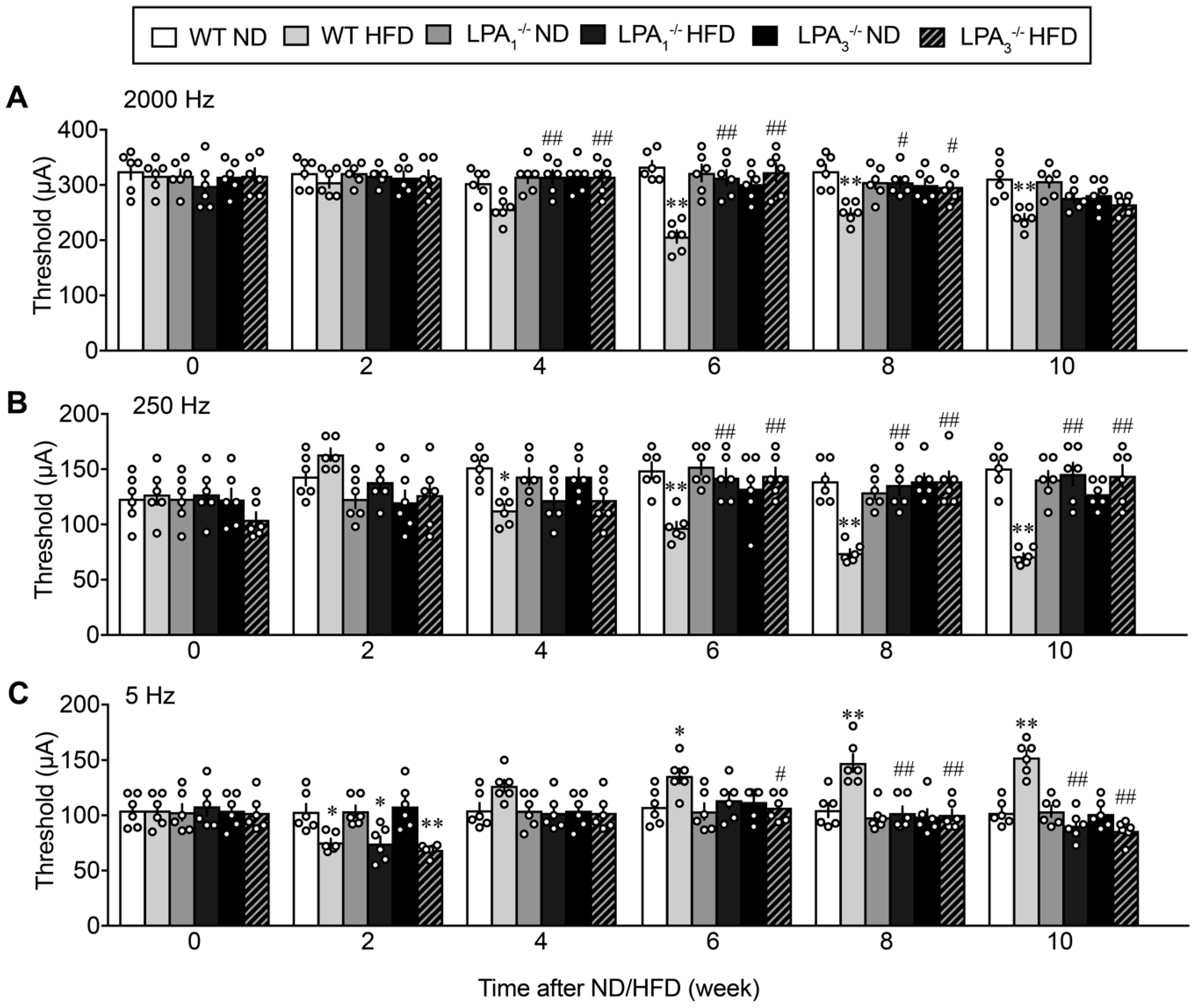
3.1. Involvement of LPA1 and LPA3 in High-Fat Diet (HFD)-Induced Abnormal Pain Behaviors
3.2. Complete Reversal of HFD-Induced Abnormal Pain Behaviors by an LPA1/3 Antagonist
3.3. Involvement of LPA1 and LPA3 in HFD-Induced Type II Diabetic Obesity
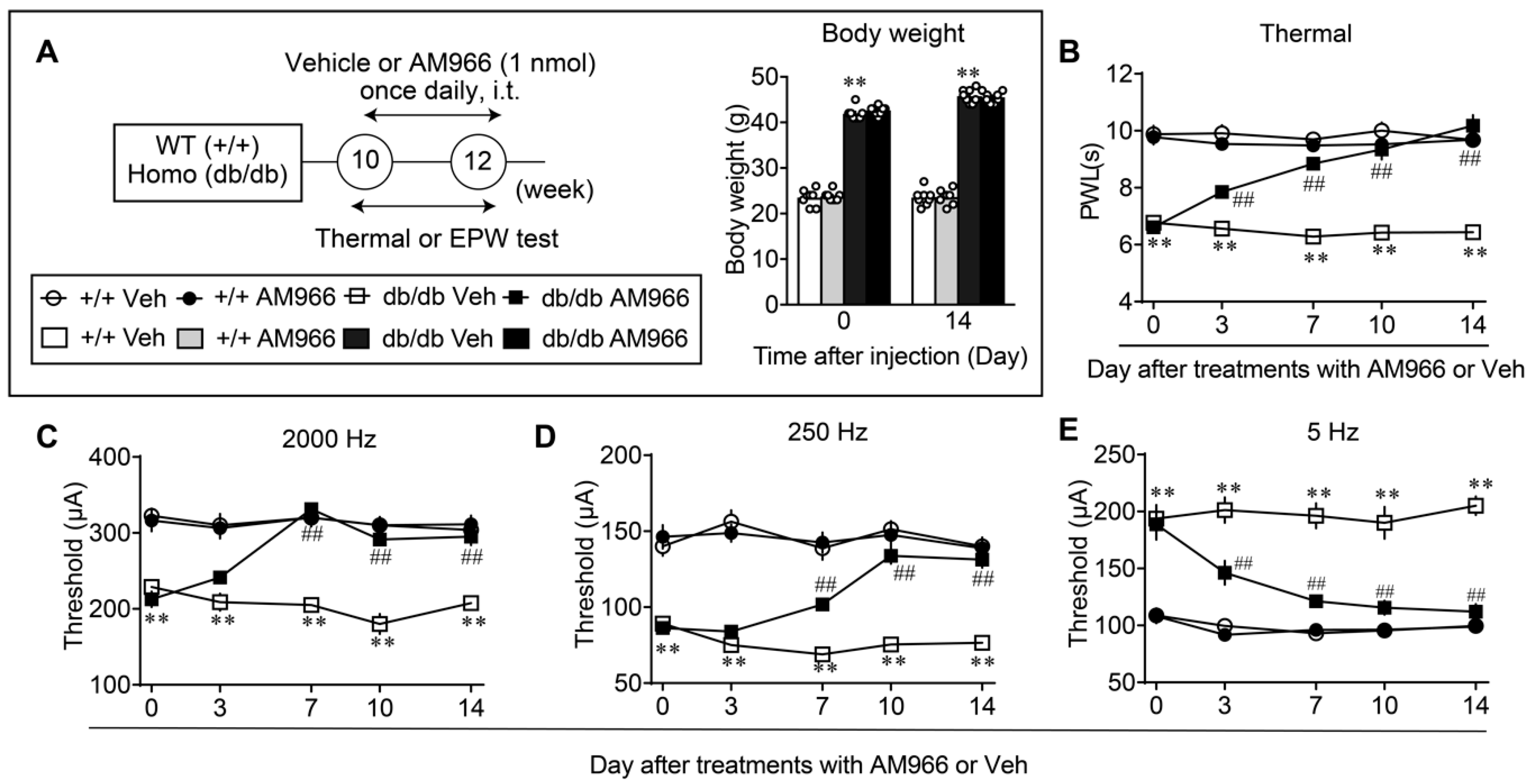
3.4. Complete Reversal of Abnormal Pain Behaviors in db/db Mice by LPA1/3 Antagonists
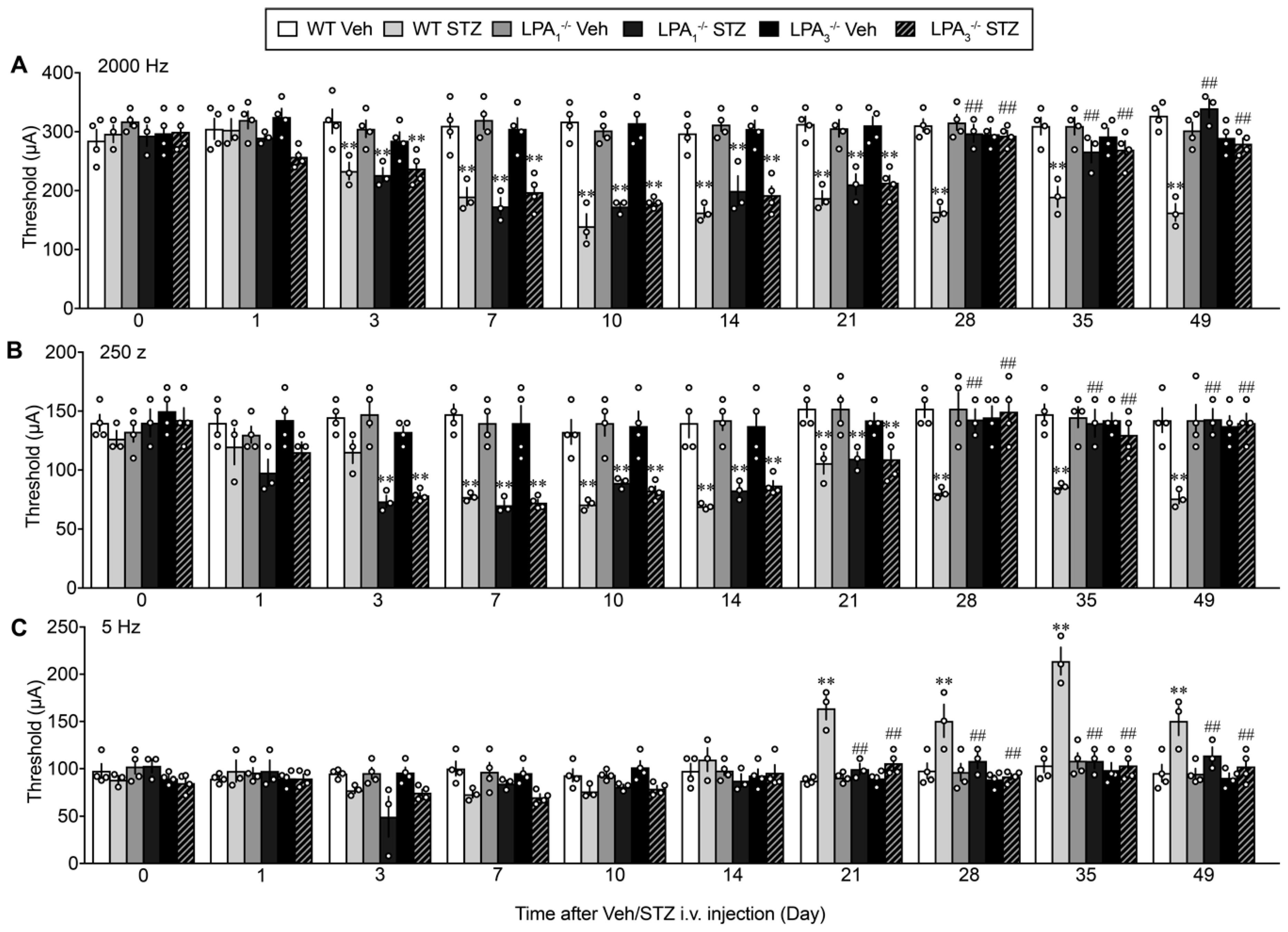
3.5. LPA1 and LPA3-Mediated Thermal Hyperalgesia in a Streptozotocin (STZ)-Induced Diabetic Mouse Model
3.6. Sensory Fiber-Specific Changes in the Nociceptive Threshold in the STZ-Model
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hansson, P. Neuropathic Pain: Clinical characteristics and diagnostic workup. Eur. J. Pain 2002, 6, 47–50. [Google Scholar] [CrossRef] [PubMed]
- Bridges, D.; Thompson, S.; Rice, A. Mechanisms of neuropathic pain. Br. J. Anaesth. 2001, 87, 12–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sah, D.W.; Ossipo, M.H.; Porreca, F. Neurotrophic factors as novel therapeutics for neuropathic pain. Nat. Rev. Drug Discov. 2003, 2, 460–472. [Google Scholar] [CrossRef] [PubMed]
- Woolf, C.J.; Mannion, R.J. Neuropathic pain: Aetiology, symptoms, mechanisms, and management. Lancet 1999, 353, 1959–1964. [Google Scholar] [CrossRef]
- Ueda, H. Molecular mechanisms of neuropathic pain–phenotypic switch and initiation mechanisms. Pharmacol. Ther. 2006, 109, 57–77. [Google Scholar] [CrossRef]
- Colloca, L.; Ludman, T.; Bouhassira, D.; Baron, R.; Dickenson, A.H.; Yarnitsky, D.; Freeman, R.; Truini, A.; Attal, N.; Finnerup, N.; et al. Neuropathic pain. Nat. Rev. Dis. Prim. 2017, 3, 17002. [Google Scholar] [CrossRef] [Green Version]
- Duby, J.J.; Campbell, R.K.; Setter, S.M.; White, J.R.; Rasmussen, K.A. Diabetic neuropathy: An intensive review. Am. J. Health Pharm. 2004, 61, 160–173. [Google Scholar] [CrossRef]
- Hicks, C.W.; Selvin, E. Epidemiology of Peripheral Neuropathy and Lower Extremity Disease in Diabetes. Curr. Diabetes Rep. 2019, 19, 86. [Google Scholar] [CrossRef]
- Jambart, S.; Ammache, Z.; Haddad, F.; Younes, A.; Hassoun, A.; Abdalla, K.; Selwan, C.A.; Sunna, N.; Wajsbrot, D.; Youseif, E. Prevalence of painful diabetic peripheral neuropathy among patients with diabetes mellitus in the Middle East region. J. Int. Med. Res. 2011, 39, 366–377. [Google Scholar] [CrossRef] [Green Version]
- Schumacher, C.; Glosner, S.E. Assessment of pain and impact of care among patients with painful diabetic peripheral neuropathy. J. Am. Pharm. Assoc. 2014, 54, 14–18. [Google Scholar] [CrossRef]
- Shahid, W.; Kumar, R.; Shaikh, A.; Kumar, S.; Jameel, R.; Fareed, S. Comparison of the Efficacy of Duloxetine and Pregabalin in Pain Relief Associated with Diabetic Neuropathy. Cureus 2019, 11, e5293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, M.; Rashid, H.; Fujita, R.; Contos, J.J.A.; Chun, J.; Ueda, H. Initiation of neuropathic pain requires lysophosphatidic acid receptor signaling. Nat. Med. 2004, 10, 712–718. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Nagai, J.; Ueda, H. Microglial activation mediates de novo lysophosphatidic acid production in a model of neuropathic pain. J. Neurochem. 2010, 115, 643–653. [Google Scholar] [CrossRef] [PubMed]
- Ueda, H. Lysophosphatidic acid signaling is the definitive mechanism underlying neuropathic pain. Pain 2017, 158, S55–S65. [Google Scholar] [CrossRef]
- Ueda, H.; Neyama, H.; Nagai, J.; Matsushita, Y.; Tsukahara, T.; Tsukahara, R. Involvement of lysophosphatidic acid–induced astrocyte activation underlying the maintenance of partial sciatic nerve injury–induced neuropathic pain. Pain 2018, 159, 2170–2178. [Google Scholar] [CrossRef] [PubMed]
- Halder, S.; Yano, R.; Chun, J.; Ueda, H. Involvement of LPA1 receptor signaling in cerebral ischemia-induced neuropathic pain. Neuroscience 2013, 235, 10–15. [Google Scholar] [CrossRef]
- Uchida, H.; Nagai, J.; Ueda, H. Lysophosphatidic acid and its receptors LPA1 and LPA3 mediate paclitaxel-induced neuropathic pain in mice. Mol. Pain 2014, 10, 71–8069. [Google Scholar] [CrossRef] [Green Version]
- Ueda, H.; Neyama, H. LPA1 receptor involvement in fibromyalgia-like pain induced by intermittent psychological stress, empathy. Neurobiol. Pain 2017, 1, 16–25. [Google Scholar] [CrossRef]
- Ueda, H.; Neyama, H.; Sasaki, K.; Miyama, C.; Iwamoto, R. Lysophosphatidic acid LPA1 and LPA3 receptors play roles in the maintenance of late tissue plasminogen activator-induced central poststroke pain in mice. Neurobiol. Pain 2018, 5, 100020. [Google Scholar] [CrossRef]
- Gil, I.G.; Zian, D.; Vázquez-Villa, H.; Hernandez-Torres, G.; Martinez, R.F.; Khiar-Fernandez, N.; Rivera, R.; Kihara, Y.; Devesa, I.; Mathivanan, S.; et al. A Novel Agonist of the Type 1 Lysophosphatidic Acid Receptor (LPA1), UCM-05194, Shows Efficacy in Neuropathic Pain Amelioration. J. Med. Chem. 2019, 63, 2372–2390. [Google Scholar] [CrossRef]
- Velasco, M.; O’Sullivan, C.A.; Sheridan, G.K. Lysophosphatidic acid receptors (LPARs): Potential targets for the treatment of neuropathic pain. Neuropharmacology 2017, 113, 608–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hylden, J.L.; Wilcox, G.L. Intrathecal morphine in mice: A new technique. Eur. J. Pharmacol. 1980, 67, 313–316. [Google Scholar] [CrossRef]
- Swaney, J.S.; Chapman, C.; Correa, L.; Stebbins, K.; Bundey, R.; Prodanovich, P.; Fagan, P.; Baccei, C.; Santini, A.; Hutchinson, J.; et al. A novel, orally active LPA1 receptor antagonist inhibits lung fibrosis in the mouse bleomycin model. Br. J. Pharmacol. 2010, 160, 1699–1713. [Google Scholar] [CrossRef] [Green Version]
- Ohta, H.; Sato, K.; Murata, N.; Damirin, A.; Malchinkhuu, E.; Kon, J.; Kimura, T.; Tobo, M.; Yamazaki, Y.; Watanabe, T.; et al. Ki16425, a Subtype-Selective Antagonist for EDG-Family Lysophosphatidic Acid Receptors. Mol. Pharmacol. 2003, 64, 994–1005. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, M. Ethical guidelines for investigations of experimental pain in conscious animals. Pain 1983, 16, 109–110. [Google Scholar] [CrossRef]
- Kilkenny, C.; Browne, W.; Cuthill, I.C.; Emerson, M.; Altman, U.G. Animal research: Reporting in vivo experiments: The ARRIVE guidelines. Br. J. Pharmacol. 2010, 160, 1577–1579. [Google Scholar] [CrossRef]
- McGrath, J.; Drummond, G.; McLachlan, E.M.; Kilkenny, C.; Wainwright, C.L. Guidelines for reporting experiments involving animals: The ARRIVE guidelines. Br. J. Pharmacol. 2010, 160, 1573–1576. [Google Scholar] [CrossRef] [Green Version]
- McGrath, J.; Lilley, E. Implementing guidelines on reporting research using animals (ARRIVE etc.): New requirements for publication in BJP. Br. J. Pharmacol. 2015, 172, 3189–3193. [Google Scholar] [CrossRef] [Green Version]
- Oike, Y.; Akao, M.; Yasunaga, K.; Yamauchi, T.; Morisada, T.; Ito, Y.; Urano, T.; Kimura, Y.; Kubota, Y.; Maekawa, H.; et al. Angiopoietin-related growth factor antagonizes obesity and insulin resistance. Nat. Med. 2005, 11, 400–408. [Google Scholar] [CrossRef]
- Akamine, T.; Kusunose, N.; Matsunaga, N.; Koyanagi, S.; Ohdo, S. Accumulation of sorbitol in the sciatic nerve modulates circadian properties of diabetes-induced neuropathic pain hypersensitivity in a diabetic mouse model. Biochem. Biophys. Res. Commun. 2018, 503, 181–187. [Google Scholar] [CrossRef]
- Rashid, H.; Inoue, M.; Bakoshi, S.; Ueda, H. Increased Expression of Vanilloid Receptor 1 on Myelinated Primary Afferent Neurons Contributes to the Antihyperalgesic Effect of Capsaicin Cream in Diabetic Neuropathic Pain in Mice. J. Pharmacol. Exp. Ther. 2003, 306, 709–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rashid, H.; Ueda, H. Nonopioid and Neuropathy-Specific Analgesic Action of the Nootropic Drug Nefiracetam in Mice. J. Pharmacol. Exp. Ther. 2002, 303, 226–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tozuka, Y.; Wada, E.; Wada, K. Diet-induced obesity in female mice leads to peroxidized lipid accumulations and impairment of hippocampal neurogenesis during the early life of their offspring. FASEB J. 2009, 23, 1920–1934. [Google Scholar] [CrossRef] [PubMed]
- Yanagita, T.; Tsuge, K.; Koga, M.; Inoue, N.; Nagao, K. Eicosapentaenoic acid-containing polar lipids from seaweed Susabinori (Pyropia yezoensis) alleviate hepatic steatosis in obese db/db mice. Arch. Biochem. Biophys. 2020, 691, 108486. [Google Scholar] [CrossRef] [PubMed]
- Hargreaves, K.; Dubner, R.; Brown, F.; Flores, C.; Joris, J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain 1988, 32, 77–88. [Google Scholar] [CrossRef]
- Uchida, H.; Ma, L.; Ueda, H. Epigenetic Gene Silencing Underlies C-Fiber Dysfunctions in Neuropathic Pain. J. Neurosci. 2010, 30, 4806–4814. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, M.; Xie, W.; Ma, L.; Ueda, H. Pharmacological switch in Aβ-fiber stimulation-induced spinal transmission in mice with partial sciatic nerve injury. Mol. Pain 2008, 4, 25. [Google Scholar] [CrossRef] [Green Version]
- Ueda, H. Peripheral mechanisms of neuropathic pain—Involvement of lysophosphatidic acid receptor-mediated demyelination. Mol. Pain 2008, 4, 11. [Google Scholar] [CrossRef] [Green Version]
- Koga, K.; Furue, H.; Rashid, H.; Takaki, A.; Katafuchi, T.; Yoshimura, M. Selective Activation of Primary Afferent Fibers Evaluated by Sine-Wave Electrical Stimulation. Mol. Pain 2005, 1, 13. [Google Scholar] [CrossRef] [Green Version]
- Szkudelski, T. The mechanism of alloxan and streptozotocin action in B cells of the rat pancreas. Physiol. Res. 2001, 50, 537–546. [Google Scholar]
- Wang, Z.; Gleichmann, H. GLUT2 in Pancreatic Islets: Crucial Target Molecule in Diabetes Induced With Multiple Low Doses of Streptozotocin in Mice. Diabetes 1998, 47, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Dyck, P.J.; Larson, T.S.; O’Brien, P.C.; Velosa, J.A. Patterns of quantitative sensation testing of hypoesthesia and hyperalgesia are predictive of diabetic polyneuropathy: A study of three cohorts. Nerve growth factor study group. Diabetes Care 2000, 23, 510–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malik, R.A. Early detection of nerve damage and repair in diabetic neuropathy. Nat. Clin. Pr. Neurol. 2008, 4, 646–647. [Google Scholar] [CrossRef]
- Harris, N.D.; Ibrahim, S.; Patel, K.A.; Selmi, F.; Radatz, M.; Ward, J.D.; Tesfaye, S.; Eaton, S.E.M. Increased sural nerve epineurial blood flow in human subjects with painful diabetic neuropathy. Diabetology 2003, 46, 934–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quattrini, C.; Harris, N.D.; Malik, R.A.; Tesfaye, S. Impaired Skin Microvascular Reactivity in Painful Diabetic Neuropathy. Diabetes Care 2007, 30, 655–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selvarajah, D.; Wilkinson, I.D.; Emery, C.J.; Shaw, P.J.; Griffiths, P.D.; Gandhi, R.; Tesfaye, S. Thalamic neuronal dysfunction and chronic sensorimotor distal symmetrical polyneuropathy in patients with type 1 diabetes mellitus. Diabetology 2008, 51, 2088–2092. [Google Scholar] [CrossRef]
- Calcutt, N.A.; Freshwater, J.D.; Mizisin, A.P. Prevention of sensory disorders in diabetic Sprague-Dawley rats by aldose reductase inhibition or treatment with ciliary neurotrophic factor. Diabetology 2004, 47, 718–724. [Google Scholar] [CrossRef] [Green Version]
- Dominguez, J.M.; Yorek, M.A.; Grant, M.B. Combination Therapies Prevent the Neuropathic, Proinflammatory Characteristics of Bone Marrow in Streptozotocin-Induced Diabetic Rats. Diabetes 2014, 64, 643–653. [Google Scholar] [CrossRef] [Green Version]
- Cameron, N.E.; Gibson, T.M.; Nangle, M.R.; Cotter, M.A. Inhibitors of Advanced Glycation End Product Formation and Neurovascular Dysfunction in Experimental Diabetes. Ann. N. Y. Acad. Sci. 2005, 1043, 784–792. [Google Scholar] [CrossRef]
- Thakur, V.; Sadanandan, J.; Chattopadhyay, M. High-Mobility Group Box 1 Protein Signaling in Painful Diabetic Neuropathy. Int. J. Mol. Sci. 2020, 21, 881. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.-S.; Kan, H.-W.; Hsieh, Y.-L. Activating transcription factor 3 modulates protein kinase C epsilon activation in diabetic peripheral neuropathy. J. Pain Res. 2019, 12, 317–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cotter, M.A.; Jack, A.M.; Cameron, N.E. Effects of the protein kinase C beta inhibitor LY333531 on neural and vascular function in rats with streptozotocin-induced diabetes. Clin. Sci. 2002, 103, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Lupachyk, S.; Shevalye, A.A.; Maksimchyk, Y.; Drel, V.; Obrosova, I.G. PARP inhibition alleviates diabetes-induced systemic oxidative stress and neural tissue 4-hydroxynonenal adduct accumulation: Correlation with peripheral nerve function. Free Radic. Biol. Med. 2011, 50, 1400–1409. [Google Scholar] [CrossRef] [Green Version]
- Pacher, P. Poly(ADP-ribose) polymerase inhibition as a novel therapeutic approach against intraepidermal nerve fiber loss and neuropathic pain associated with advanced diabetic neuropathy: A commentary on “PARP Inhibition or gene deficiency counteracts intraepidermal nerve fiber loss and neuropathic pain in advanced diabetic neuropathy”. Free Radic. Biol. Med. 2008, 44, 969–971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cameron, N.E.; Jack, A.M.; Cotter, M.A.; Cameron, N.E. Effect of α-lipoic acid on vascular responses and nociception in diabetic rats. Free Radic. Biol. Med. 2001, 31, 125–135. [Google Scholar] [CrossRef]
- Cameron, N.E.; Tuck, Z.; McCabe, L.; Cotter, M.A. Effect of the hydroxyl radical scavenger, dimethylthiourea, on peripheral nerve tissue perfusion, conduction velocity and nociception in experimental diabetes. Diabetology 2001, 44, 1161–1169. [Google Scholar] [CrossRef] [Green Version]
- Kahya, M.C.; Nazıroğlu, M.; Övey, I.S. Modulation of Diabetes-Induced Oxidative Stress, Apoptosis, and Ca2+ Entry Through TRPM2 and TRPV1 Channels in Dorsal Root Ganglion and Hippocampus of Diabetic Rats by Melatonin and Selenium. Mol. Neurobiol. 2016, 54, 2345–2360. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, N.; Helmstädter, J.; Rojas, D.R.; Bali, K.K.; Gangadharan, V.; Kuner, R. Evoked hypoalgesia is accompanied by tonic pain and immune cell infiltration in the dorsal root ganglia at late stages of diabetic neuropathy in mice. Mol. Pain 2018, 14, 1744806918817975. [Google Scholar] [CrossRef]
- Obrosova, I.G.; Xu, W.; Lyzogubov, V.V.; Ilnytska, O.; Mashtalir, N.; Vareniuk, I.; Pavlov, I.A.; Zhang, J.; Slusher, B.; Drel, V. PARP inhibition or gene deficiency counteracts intraepidermal nerve fiber loss and neuropathic pain in advanced diabetic neuropathy. Free Radic. Biol. Med. 2008, 44, 972–981. [Google Scholar] [CrossRef] [Green Version]
- Pabbidi, R.M.; Yu, S.-Q.; Peng, S.; Khardori, R.; E Pauza, M.; Premkumar, L.S. Influence of TRPV1 on diabetes-induced alterations in thermal pain sensitivity. Mol. Pain 2008, 4, 9. [Google Scholar] [CrossRef] [Green Version]
- Prnova, M.S.; Kovacikova, L.; Svik, K.; Bezek, S.; Elmazoğlu, Z.; Karasu, C.; Štefek, M. Triglyceride-lowering effect of the aldose reductase inhibitor cemtirestat—Another factor that may contribute to attenuation of symptoms of peripheral neuropathy in STZ-diabetic rats. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2019, 393, 651–661. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, S.K.R.; Smith, D.R.; Saleh, A.; Schapansky, J.; Marquez, A.; Gomes, S.; Akude, E.; Morrow, D.; Calcutt, N.A.; Fernyhough, P. Impaired adenosine monophosphate-activated protein kinase signalling in dorsal root ganglia neurons is linked to mitochondrial dysfunction and peripheral neuropathy in diabetes. Brain 2012, 135, 1751–1766. [Google Scholar] [CrossRef] [PubMed]
- Davidson, E.P.; Coppey, L.J.; Kleinschmidt, T.L.; Oltman, C.L.; Yorek, M.A. Vascular and Neural Dysfunctions in Obese Zucker Rats: Effect of AVE7688. Exp. Diabetes Res. 2009, 2009, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obrosova, I.G.; Ilnytska, O.; Lyzogubov, V.V.; Pavlov, I.A.; Mashtalir, N.; Nadler, J.L.; Drel, V. High-Fat Diet Induced Neuropathy of Pre-Diabetes and Obesity: Effects of “Healthy” Diet and Aldose Reductase Inhibition. Diabetes 2007, 56, 2598–2608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, T.-J.S.; Zhang, M.-D.; Zeberg, H.; Nilsson, J.; Grünler, J.; Liu, S.-X.; Xiang, Q.; Persson, J.; Fried, K.J.; Catrina, S.-B.; et al. Coenzyme Q10 prevents peripheral neuropathy and attenuates neuron loss in the db-/db- mouse, a type 2 diabetes model. Proc. Natl. Acad. Sci. USA 2012, 110, 690–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ørstavik, K.; Namer, B.; Schmidt, R.; Schmelz, M.; Hilliges, M.; Weidner, C.; Carr, R.W.; Handwerker, H.; Jørum, E.; Torebjork, H.E. Abnormal Function of C-Fibers in Patients with Diabetic Neuropathy. J. Neurosci. 2006, 26, 11287–11294. [Google Scholar] [CrossRef] [Green Version]
- Inoue, M.; Ma, L.; Aoki, J.; Ueda, H. Simultaneous stimulation of spinal NK1 and NMDA receptors produces LPC which undergoes ATX-mediated conversion to LPA, an initiator of neuropathic pain. J. Neurochem. 2008, 107, 1556–1565. [Google Scholar] [CrossRef]
- Kang, S.; Han, J.; Song, S.Y.; Kim, W.-S.; Shin, S.; Kim, J.H.; Ahn, H.; Jeong, J.-H.; Hwang, S.-J.; Sung, J.-H. Lysophosphatidic acid increases the proliferation and migration of adipose-derived stem cells via the generation of reactive oxygen species. Mol. Med. Rep. 2015, 12, 5203–5210. [Google Scholar] [CrossRef] [Green Version]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ueda, H.; Neyama, H.; Matsushita, Y. Lysophosphatidic Acid Receptor 1- and 3-Mediated Hyperalgesia and Hypoalgesia in Diabetic Neuropathic Pain Models in Mice. Cells 2020, 9, 1906. https://doi.org/10.3390/cells9081906
Ueda H, Neyama H, Matsushita Y. Lysophosphatidic Acid Receptor 1- and 3-Mediated Hyperalgesia and Hypoalgesia in Diabetic Neuropathic Pain Models in Mice. Cells. 2020; 9(8):1906. https://doi.org/10.3390/cells9081906
Chicago/Turabian StyleUeda, Hiroshi, Hiroyuki Neyama, and Yosuke Matsushita. 2020. "Lysophosphatidic Acid Receptor 1- and 3-Mediated Hyperalgesia and Hypoalgesia in Diabetic Neuropathic Pain Models in Mice" Cells 9, no. 8: 1906. https://doi.org/10.3390/cells9081906
APA StyleUeda, H., Neyama, H., & Matsushita, Y. (2020). Lysophosphatidic Acid Receptor 1- and 3-Mediated Hyperalgesia and Hypoalgesia in Diabetic Neuropathic Pain Models in Mice. Cells, 9(8), 1906. https://doi.org/10.3390/cells9081906