Clinical and Molecular Characterization of Classical-Like Ehlers-Danlos Syndrome Due to a Novel TNXB Variant


 ,
,  ,
,  , , and
, , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethical Compliance
2.2. Cell Culture
2.3. Molecular Analysis
2.4. RNA Extraction and Quantitative Real-Time PCR
2.5. Collagen Biochemical, Ultrastructural and Immunofluorescence Microscopy Studies
3. Results
3.1. Case Report
3.2. Molecular and Biochemical Findings
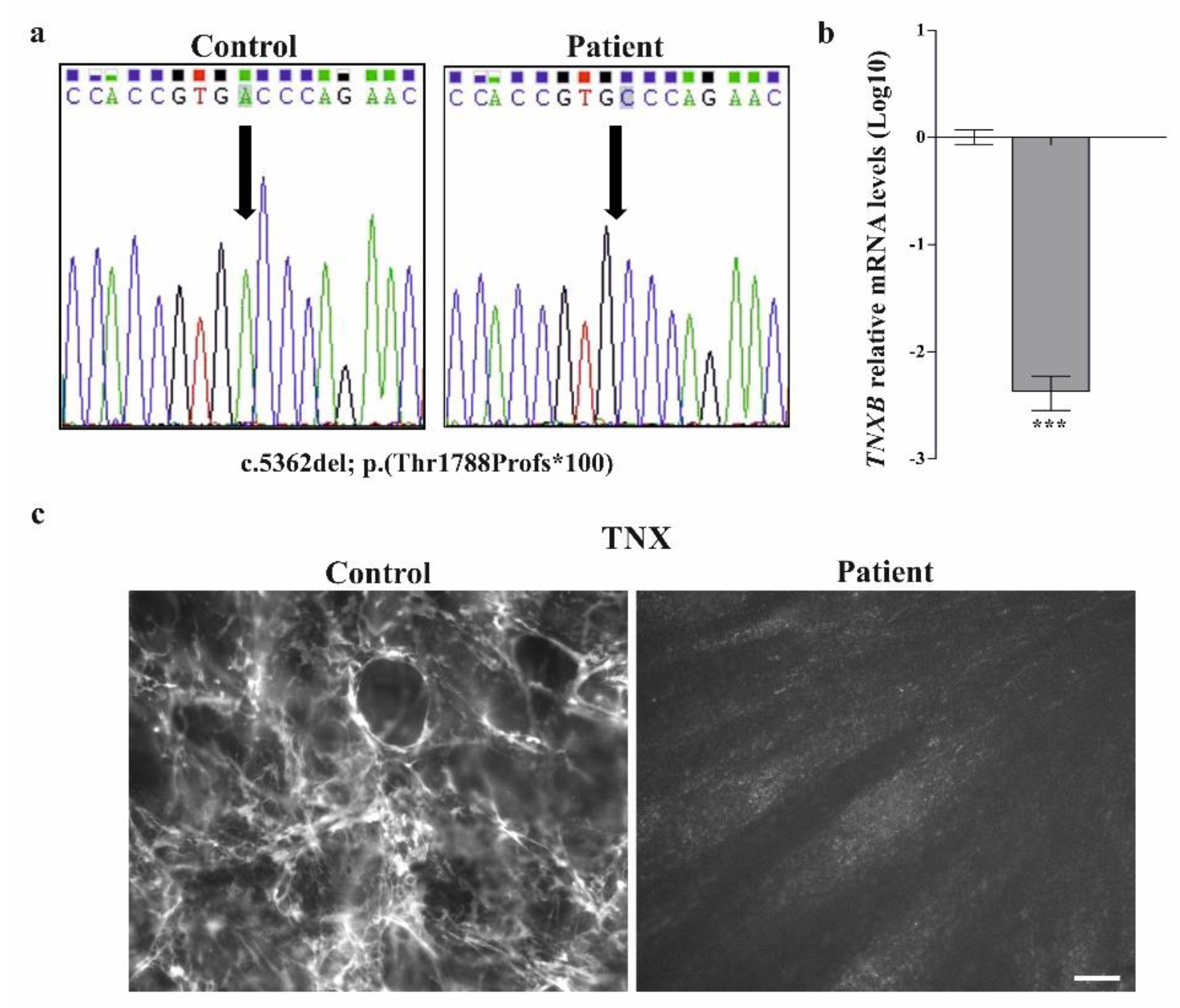
3.2.1. Molecular Analysis
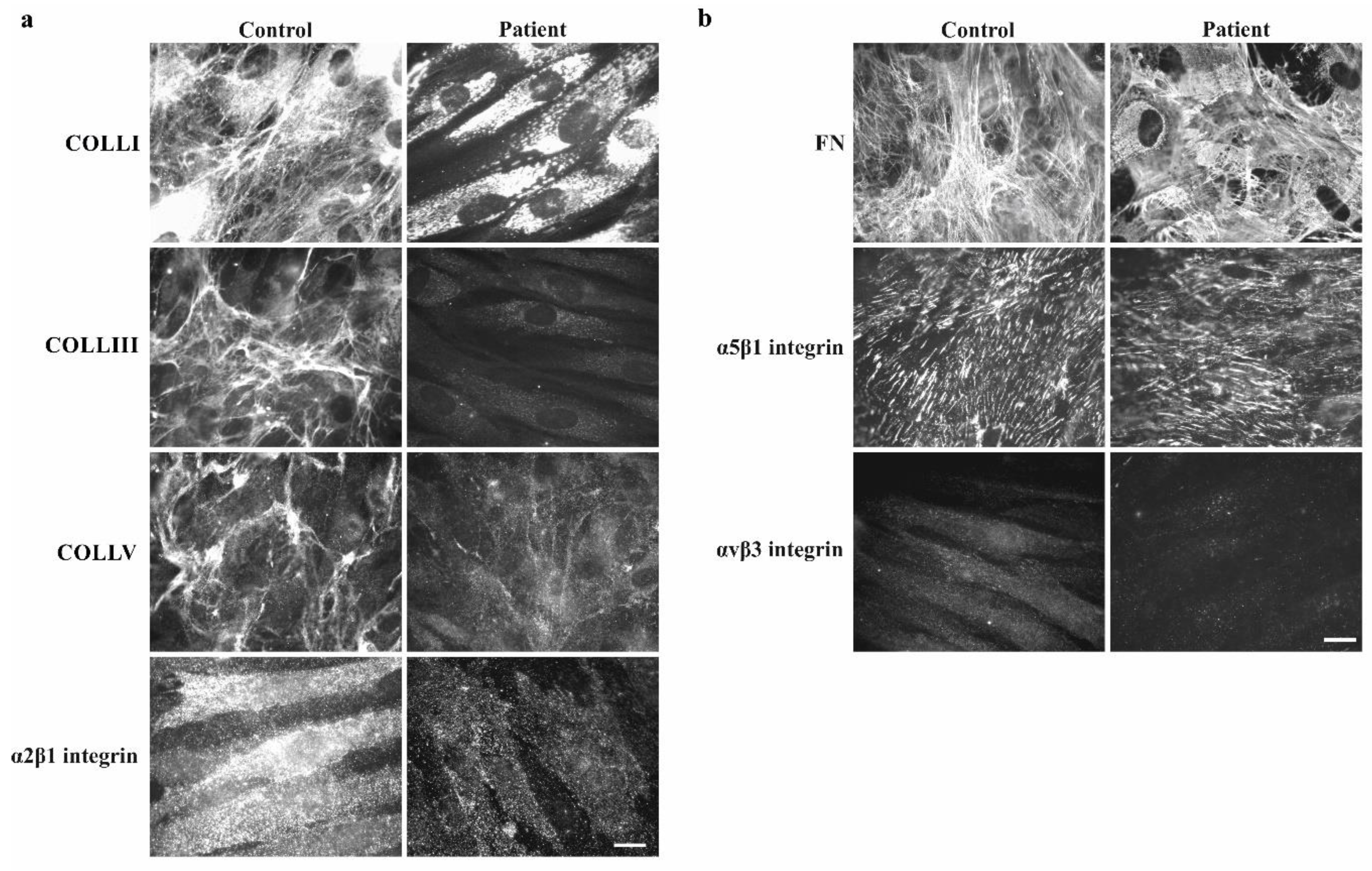
3.2.2. Collagen Biochemical and Immunofluorescent Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Brady, A.F.; Demirdas, S.; Fournel-Gigleux, S.; Ghali, N.; Giunta, C.; Kapferer-Seebacher, I.; Kosho, T.; Mendoza-Londono, R.; Pope, M.F.; van Damme, T.; et al. The Ehlers–Danlos syndromes, rare types. Am. J. Med. Genet. 2017, 175, 70–115. [Google Scholar] [CrossRef] [PubMed]
- Chiquet-Ehrismann, R.; Tucker, R.P. Tenascins and the importance of adhesion modulation. Cold Spring Harb. Perspect. Biol. 2011, 3, a004960. [Google Scholar] [CrossRef] [PubMed]
- Elefteriou, F.; Exposito, J.Y.; Garrone, R.; Lethias, C. Binding of tenascin-X to decorin. FEBS Lett. 2001, 495, 44–47. [Google Scholar] [CrossRef] [Green Version]
- Zweers, M.C.; van Vlijmen-Willems, I.M.; van Kuppevelt, T.H.; Mecham, R.P.; Steijlen, P.M.; Bristow, J.; Schalkwijk, J. Deficiency of tenascin-X causes abnormalities in dermal elastic fiber morphology. J. Investig. Dermatol. 2004, 122, 885–891. [Google Scholar] [CrossRef] [PubMed]
- Bristow, J.; Carey, W.; Egging, D.; Schalkwijk, J. Tenascin-X, collagen, elastin, and the Ehlers–Danlos syndrome. Am. J. Med. Genet. 2005, 139, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.I.; Takayama, N.; Ohnishi, J.; Ohnishi, E.; Shirayoshi, Y.; Nakatsuji, N.; Ariga, H. Tumour invasion and metastasis are promoted in mice deficient in tenascin-X. Genes Cells 2001, 6, 1101–1111. [Google Scholar] [CrossRef]
- Minamitani, T.; Ariga, H.; Matsumoto, K.I. Deficiency of tenascin-X causes a decrease in the level of expression of type VI collagen. Exp. Cell Res. 2004, 297, 49–60. [Google Scholar] [CrossRef]
- Burch, G.H.; Gong, Y.; Liu, W.; Dettman, R.W.; Curry, C.J.; Smith, L.; Miller, W.L.; Bristow, J. Tenascin-X deficiency is associated with Ehlers-Danlos syndrome. Nature 1997, 17, 104–108. [Google Scholar] [CrossRef]
- Schalkwijk, J.; Zweers, M.C.; Steijlen, P.M.; Dean, W.B.; Taylor, G.; van Vlijmen, I.M.; van Haren, B.; Miller, W.L.; Bristow, J. A recessive form of Ehlers-Danlos syndrome caused by tenascin-X deficiency. N. Engl. J. Med. 2001, 345, 1167–1175. [Google Scholar] [CrossRef]
- Mao, J.R.; Taylor, G.; Dean, W.B.; Wagner, D.R.; Afzal, V.; Lotz, J.C.; Rubin, E.M.; Bristow, J. Tenascin-X deficiency mimics Ehlers-Danlos syndrome in mice through alteration of collagen deposition. Nat. Genet. 2002, 30, 421–425. [Google Scholar] [CrossRef]
- Yung, Y.C. Molecular genetics of the human MHC complement gene cluster. Exp. Clin. Immunogenet. 1998, 15, 213–230. [Google Scholar]
- Lindert, U.; Gnoli, M.; Maioli, M.; Bedeschi, M.F.; Sangiorgi, L.; Rohrbach, M.; Giunta, C. Insight into the pathology of a COL1A1 signal peptide heterozygous mutation leading to severe osteogenesis imperfecta. Calcif. Tissue Int. 2018, 102, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Zoppi, N.; Gardella, R.; de Paepe, A.; Barlati, S.; Colombi, M. Human fibroblasts with mutations in COL5A1 and COL3A1 genes do not organize collagens and fibronectin in the extracellular matrix, down-regulate α2β1 integrin, and recruit α vβ3 instead of α5β1 integrin. J. Biol. Chem. 2004, 279, 18157–18168. [Google Scholar] [CrossRef] [PubMed]
- Zoppi, N.; Barlati, S.; Colombi, M. FAK-independent αvβ3 integrin-EGFR complexes rescue from anoikis matrix-defective fibroblasts. Biochim. Biophys. Acta Mol. Cell Res. 2008, 1783, 1177–1188. [Google Scholar] [CrossRef] [PubMed]
- Chiarelli, N.; Carini, G.; Zoppi, N.; Dordoni, C.; Ritelli, M.; Venturini, M.; Castori, M.; Colombi, M. Transcriptome-wide expression profiling in skin fibroblasts of patients with joint hypermobility syndrome/ehlers-danlos syndrome hypermobility type. PLoS ONE 2016, 11, e0161347. [Google Scholar] [CrossRef]
- Zoppi, N.; Chiarelli, N.; Binetti, S.; Ritelli, M.; Colombi, M. Dermal fibroblast-to-myofibroblast transition sustained by α vß3integrin-ILK-Snail1/Slug signaling is a common feature for hypermobile Ehlers-Danlos syndrome and hypermobility spectrum disorders. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1010–1023. [Google Scholar] [CrossRef]
- Zoppi, N.; Chiarelli, N.; Ritelli, M.; Colombi, M. Multifaced roles of the αvβ3 integrin in Ehlers–Danlos and arterial tortuosity syndromes’ dermal fibroblasts. Int. J. Mol. Sci. 2018, 19, 982. [Google Scholar] [CrossRef]
- Lindor, N.M.; Bristow, J. Tenascin-X deficiency in autosomal recessive Ehlers-Danlos syndrome. Am. J. Med. Genet. 2005, 135, 75–80. [Google Scholar] [CrossRef]
- Pénisson-Besnier, I.; Allamand, V.; Beurrier, P.; Martin, L.; Schalkwijk, J.; van Vlijmen-Willems, I.; Gartioux, C.; Malfait, F.; Syx, D.; Marcorelles, P.; et al. Compound heterozygous mutations of the TNXB gene cause primary myopathy. Neuromuscul. Disord. 2013, 23, 664–669. [Google Scholar] [CrossRef]
- Hendriks, A.G.; Voermans, N.C.; Schalkwijk, J.; Hamel, B.C.; van Rossum, M.M. Well-defined clinical presentation of Ehlers-Danlos syndrome in patients with tenascin-X deficiency: A report of four cases. Clin. Dysmorphol. 2012, 21, 15–18. [Google Scholar] [CrossRef]
- Voermans, N.C.; van Alfen, N.; Pillen, S.; Lammens, M.; Schalkwijk, J.; Zwarts, M.J.; van Rooij, I.A.; Hamel, B.C.J.; van Engelen, B.G. Neuromuscular involvement in various types of Ehlers-Danlos syndrome. Ann. Neurol. 2009, 65, 687–697. [Google Scholar] [CrossRef] [PubMed]
- Sakiyama, T.; Kubo, A.; Sasaki, T.; Yamada, T.; Yabe, N.; Matsumoto, K.I.; Futei, Y. Recurrent gastrointestinal perforation in a patient with Ehlers-Danlos syndrome due to tenascin-X deficiency. J. Dermatol. 2015, 42, 511–514. [Google Scholar] [CrossRef] [PubMed]
- Demirdas, S.; Dulfer, E.; Robert, L.; Kempers, M.; van Beek, D.; Micha, D.; van Engelen, B.G.; Hamel, B.; Schalkwijk, J.; Maugeri, A.; et al. Recognizing the tenascin-X deficient type of Ehlers–Danlos syndrome: A cross-sectional study in 17 patients. Clin. Genet. 2017, 91, 411–425. [Google Scholar] [CrossRef]
- O’Connell, M.; Burrows, N.P.; van Vlijmen-Willems, M.J.J.; Clark, S.M.; Schalkwijk, J. Tenascin-X deficiency and Ehlers-Danlos syndrome: A case report and review of the literature. Br. J. Dermatol. 2010, 163, 1340–1345. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Perritt, A.F.; Morissette, R.; Dreiling, J.L.; Bohn, M.F.; Mallappa, A.; Xu, Z.; Quezado, M.; Merke, D.P. Ehlers-Danlos syndrome caused by biallelic TNXB variants in patients with congenital adrenal hyperplasia. Hum. Mutat. 2016, 37, 893–897. [Google Scholar] [CrossRef] [PubMed]
- Mackenroth, L.; Fischer-Zirnsak, B.; Egerer, J.; Hecht, J.; Kallinich, T.; Stenzel, W.; Spors, B.; von Moers, A.; Mundlos, S.; Gerhold, K.; et al. An overlapping phenotype of osteogenesis imperfecta and Ehlers-Danlos syndrome due to a heterozygous mutation in COL1A1 and biallelic missense variants in TNXB identified by whole exome sequencing. Am. J. Med. Genet. 2016, 170, 1080–1085. [Google Scholar] [CrossRef] [PubMed]
- Egging, D.; van Vlijmen-Willems, I.; van Tongeren, T.; Schalkwijk, J.; Peeters, A. Wound healing in tenascin-X deficient mice suggests that tenascin-X is involved in matrix maturation rather than matric deposition. Connect. Tissue Res. 2007, 48, 93–98. [Google Scholar] [CrossRef]
- Burch, G.H.; Bedolli, M.A.; McDonough, S.; Rosenthal, S.M.; Bristow, J. Embryonic expression of tenascin-X suggests a role in limb, muscle and heart development. Dev. Dyn. 1995, 203, 491–504. [Google Scholar] [CrossRef]
- Naoum, J.J.; Hunter, G.C.; Woodside, K.J.; Chen, C. Current advances in the pathogenesis of varicose veins. J. Surg. Res. 2007, 141, 311–316. [Google Scholar] [CrossRef]
- Ikuta, T.; Ariga, H.; Matsumoto, K. Extracellular matrix tenascin-X in combination with vascular endothelial growth factor B enhances endothelial cell proliferation. Genes Cells 2000, 5, 913–927. [Google Scholar] [CrossRef]
- Ishitsuka, T.; Ikuta, T.; Ariga, H.; Matsumoto, K. Serum tenascin-X binds to vascular endothelial growth factor. Biol. Pharm. Bull. 2009, 32, 1004–1011. [Google Scholar] [CrossRef] [PubMed]
- Formenti, A.M.; Doga, M.; Frara, S.; Ritelli, M.; Colombi, M.; Banfi, G.; Giustina, A. Skeletal fragility: An emerging complication of Ehlers-Danlos syndrome. Endocrine 2019, 63, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Kajitani, N.; Yamada, T.; Kawakami, K.; Matsumoto, K.I. TNX deficiency results in bone loss due to an increase in multinucleated osteoclasts. Biochem. Biophys. Res. Commun. 2019, 512, 659–664. [Google Scholar] [CrossRef] [PubMed]
- Voermans, N.C.; Verrijp, K.; Eshuis, L.; Balemans, M.C.M.; Egging, D.; Sterrenburg, E.; van Rooij, I.A.L.M.; van der Laak, J.A.W.M.; Schalkwijk, J.; Lammens, M.; et al. Mild muscular features in tenascin-X knockout mice, a model of Ehlers-Danlos syndrome. Connect. Tissue Res. 2011, 52, 422–432. [Google Scholar] [CrossRef]
- Kirschner, J.; Hausser, I.; Zou, Y.; Schreiber, G.; Christen, H.J.; Brown, S.C.; Anton-Lamprecht, I.; Muntoni, F.; Hanefeld, F.; Bönnemann, C.G. Ullrich congenital muscular dystrophy: Connective tissue abnormalities in the skin support overlap with Ehlers–Danlos syndromes. Am. J. Hum. Genet. 2004, 132, 296–301. [Google Scholar] [CrossRef]
- Besselink-Lobanova, A.; Maandag, N.J.; Voermans, N.C.; van der Heijden, H.F.; van der Hoeven, J.G.; Heunks, L.M. Trachea rupture in tenascin-X deficient type Ehlers-Danlos syndrome. Anesthesiology 2010, 113, 746–749. [Google Scholar] [CrossRef]
- Peeters, A.C.T.; Kucharekova, M.; Timmermans, J.; van den Berkmortel, F.W.P.J.; Boers, G.H.J.; Novakova, I.R.O.; Egging, D.F.; den Heijer, M.; Schalkwijk, J. A clinical and cardiovascular survey of Ehlers-Danlos syndrome patients with complete deficiency of tenascin-X. Neth. J. Med. 2004, 62, 160–162. [Google Scholar]
- Imanaka-Yoshida, K.; Matsumoto, K. Multiple roles of tenascins in homeostasis and pathophysiology of aorta. Ann. Vasc. Dis. 2018, 11, 169–180. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Present Patient | clEDS* | Total (%) | |
|---|---|---|---|
| Major criteria | |||
| Skin hyperextensibility (with velvety skin and absence of atrophic scarring) | + | 19/19 | 20/20 (100) |
| GJH (Beighton Score > 5/9) (+/− recurrent dislocation) | − | 15/18 | 15/19 (79) |
| Easy bruising | + | 19/19 | 20/20 (100) |
| Minor Criteria | |||
| Articular (sub)luxation | + | 17/18 | 18/19 (95) |
| Feet deformities (broad/short feet, brachydactyly, pes planus, hallux valgus) | + | 14/18 | 15/19 (79) |
| Hand deformities (acrogeric hands, mallet fingers, clinodactyly and brachydactyly) | + | 4/17 | 5/18 (28) |
| Mild proximal and distal muscle weakness | + | 9/19 | 10/20 (50) |
| Polyneuropathy | NA | 7/18 | 7/18 (39) |
| Edema in the legs in absence of cardiac failure | + | 4/17 | 5/18 (28) |
| Other features | |||
| Congenital joint dislocation | + | 1/17 | 2/18 (11) |
| Delayed wound healing | + | 7/18 | 8/19 (42) |
| Vaginal/uterus/rectal prolapse | − | 5/17 | 5/18 (28) |
| Inguinal/umbilical/wound herniation | + | 4/19 | 5/20 (25) |
| Varicose veins | + | 3/17 | 4/18 (22) |
| Piezogenic papulae | + | 13/18 | 14/19 (74) |
| High arched/narrow palate +/− dental crowding | + | 4/17 | 5/18 (28) |
| Refractive error | + | 8/17 | 9/18 (50) |
| Mitral valve abnormalities | − | 4/19 | 4/20 (20) |
| Diverticulosis/diverticulitis | + | 4/18 | 5/19 (26) |
| Spontaneous bowel perforation | + | 1/18 | 2/19 (11) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rymen, D.; Ritelli, M.; Zoppi, N.; Cinquina, V.; Giunta, C.; Rohrbach, M.; Colombi, M. Clinical and Molecular Characterization of Classical-Like Ehlers-Danlos Syndrome Due to a Novel TNXB Variant. Genes 2019, 10, 843. https://doi.org/10.3390/genes10110843
Rymen D, Ritelli M, Zoppi N, Cinquina V, Giunta C, Rohrbach M, Colombi M. Clinical and Molecular Characterization of Classical-Like Ehlers-Danlos Syndrome Due to a Novel TNXB Variant. Genes. 2019; 10(11):843. https://doi.org/10.3390/genes10110843
Chicago/Turabian StyleRymen, Daisy, Marco Ritelli, Nicoletta Zoppi, Valeria Cinquina, Cecilia Giunta, Marianne Rohrbach, and Marina Colombi. 2019. "Clinical and Molecular Characterization of Classical-Like Ehlers-Danlos Syndrome Due to a Novel TNXB Variant" Genes 10, no. 11: 843. https://doi.org/10.3390/genes10110843
APA StyleRymen, D., Ritelli, M., Zoppi, N., Cinquina, V., Giunta, C., Rohrbach, M., & Colombi, M. (2019). Clinical and Molecular Characterization of Classical-Like Ehlers-Danlos Syndrome Due to a Novel TNXB Variant. Genes, 10(11), 843. https://doi.org/10.3390/genes10110843