Genistein Modulates Signaling Pathways and Targets Several Epigenetic Markers in HeLa Cells

 , and
, and 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Cell Line and Cell Culture
2.2. Preparation of Genistein
2.3. Expression Analysis of Various Genes Involved in Tumorigenesis and Cancer Related Pathways
2.4. DNA Methyltransferase Activity Assay
2.5. Histone Deacetylase Activity Assay
2.6. Histone Methyltransferase-H3K9 Activity Assay
2.7. Expression Analysis of the Genes Involved in Chromatin Modification
2.8. Global DNA Methylation Assay
2.9. Detection of Promoter Methylation Using Methylation Array
2.10. Statistical Analysis of Experimental Data
2.11. In silico Network Analysis of Target Genes
2.12. Gene Function and Pathway Enrichment Analysis
3. Results
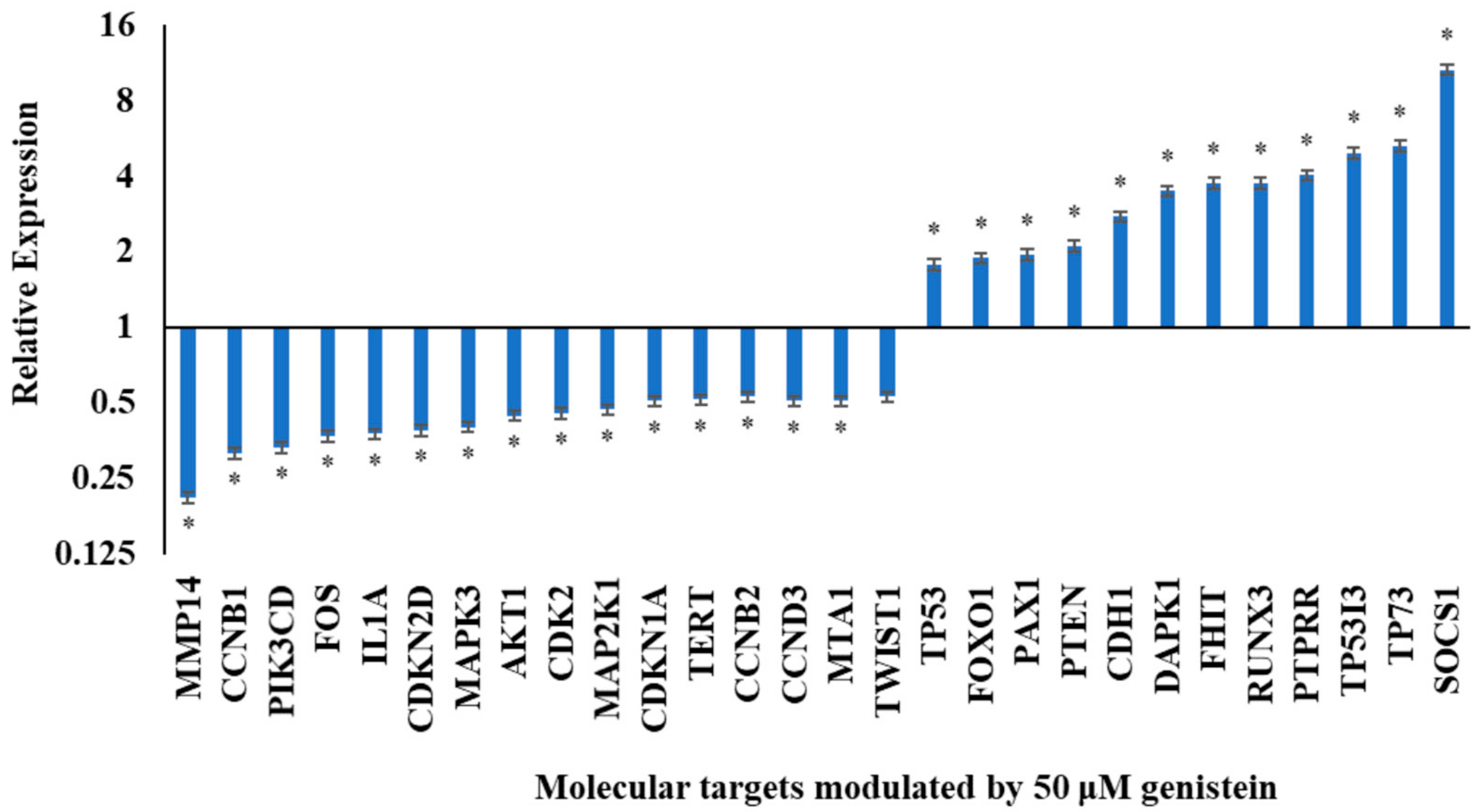
3.1. Genistein Modulates the Expression of Genes Involved in Cell Cycle Regulation, Migration, Signaling Pathways and Inflammation
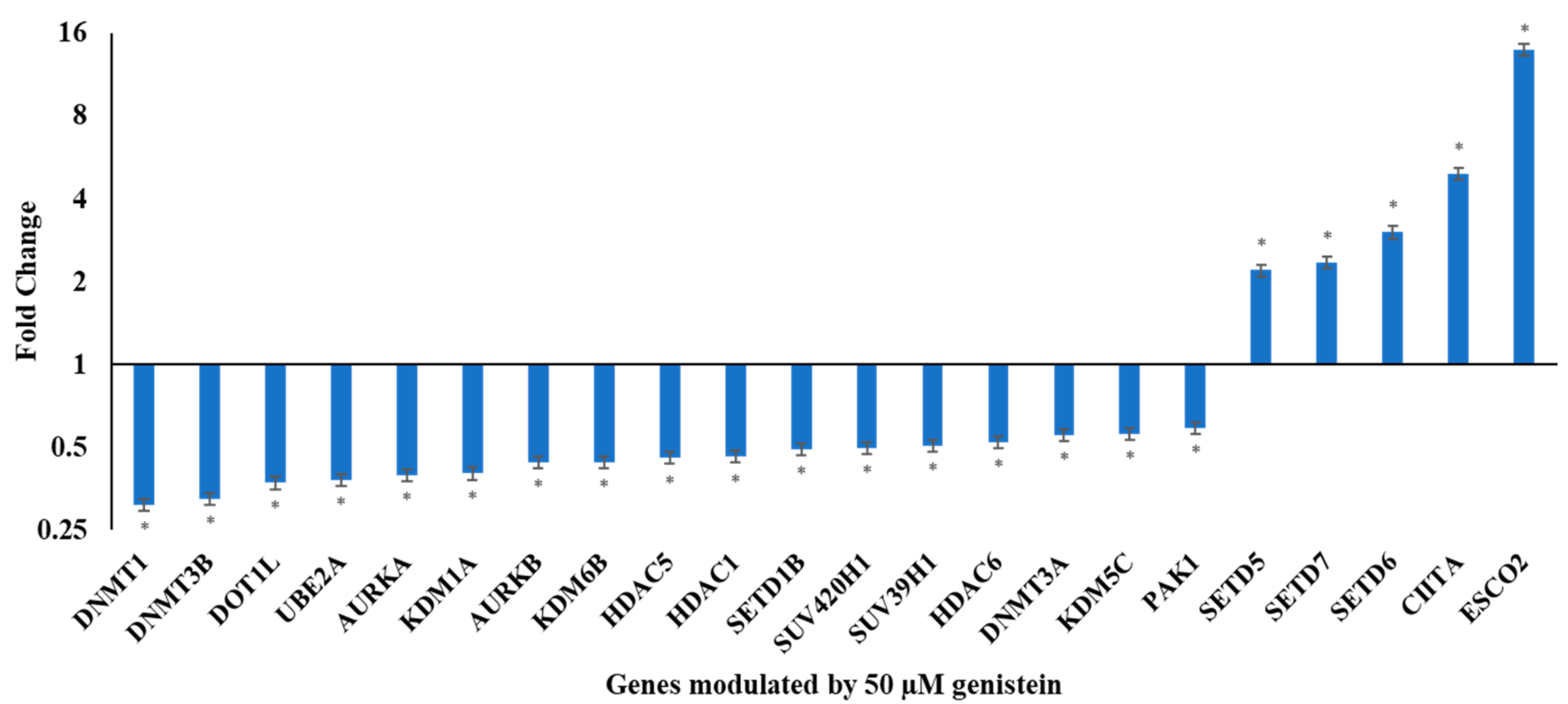
3.2. Genistein Modulates the Expression of Various Chromatin Modifiers Involved in the Epigenetic Pathway
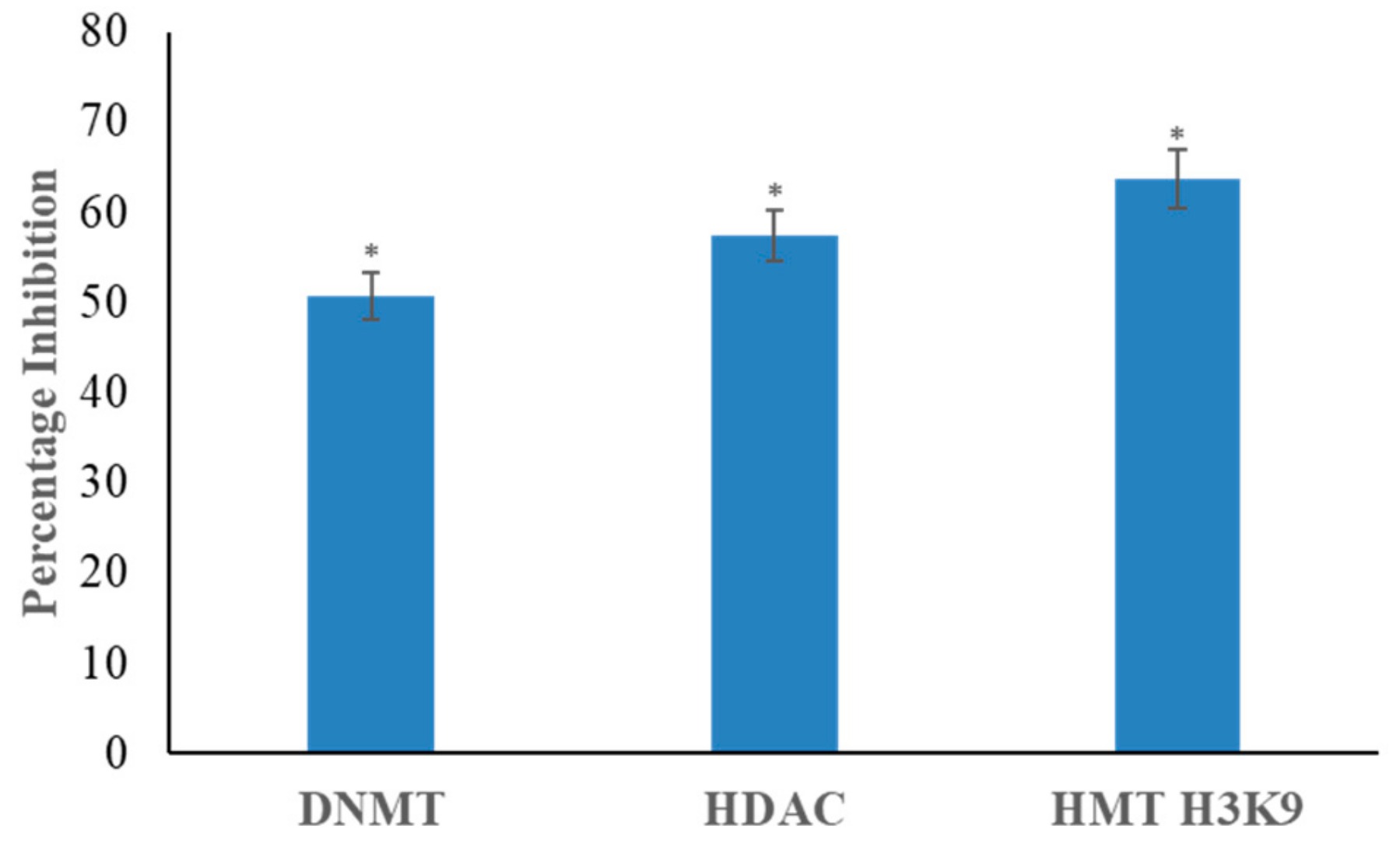
3.3. Genistein Inhibits DNMT, HDAC and HMT H3K9 Activity
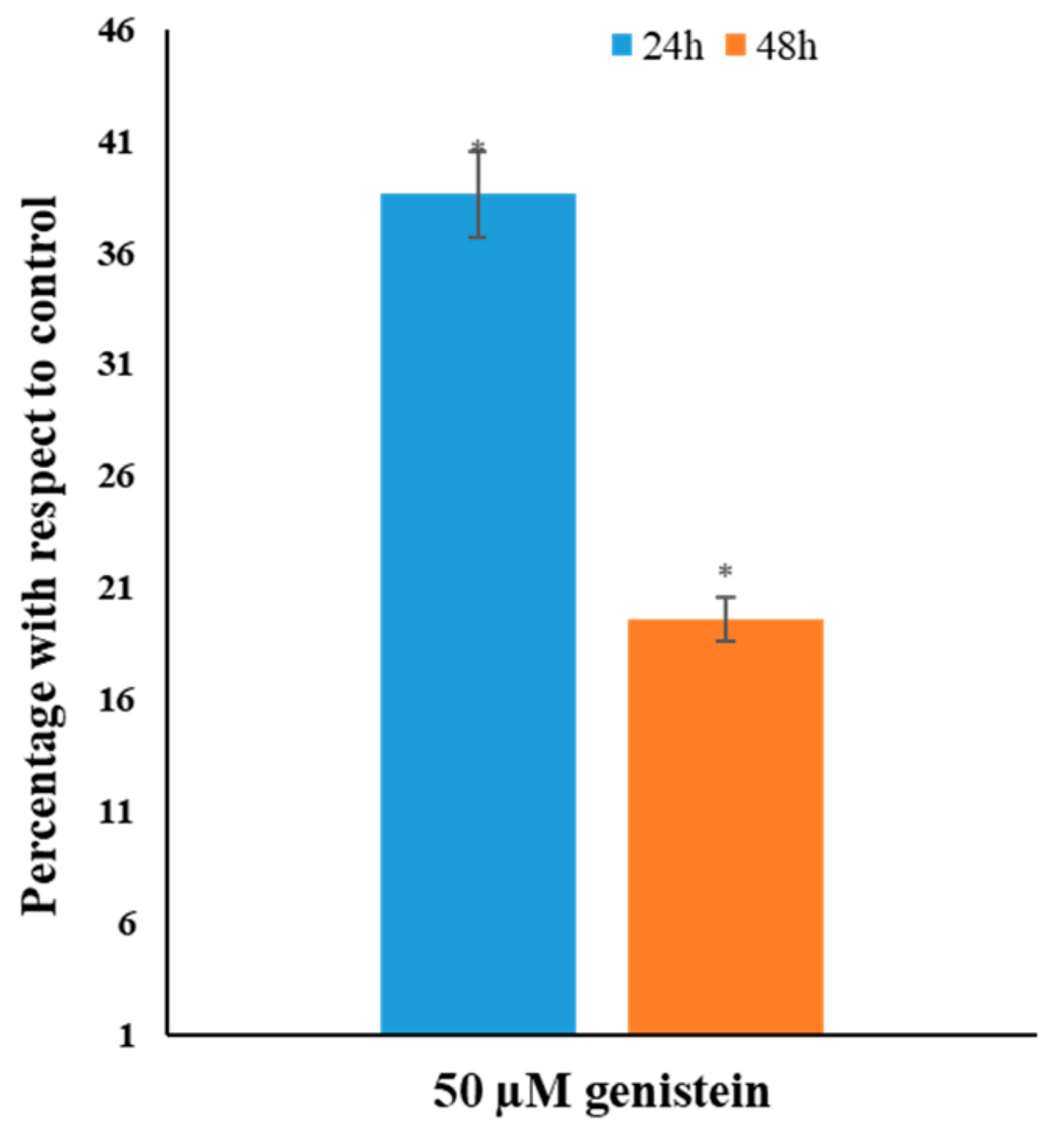
3.4. Genistein Decreases Global DNA Methylation
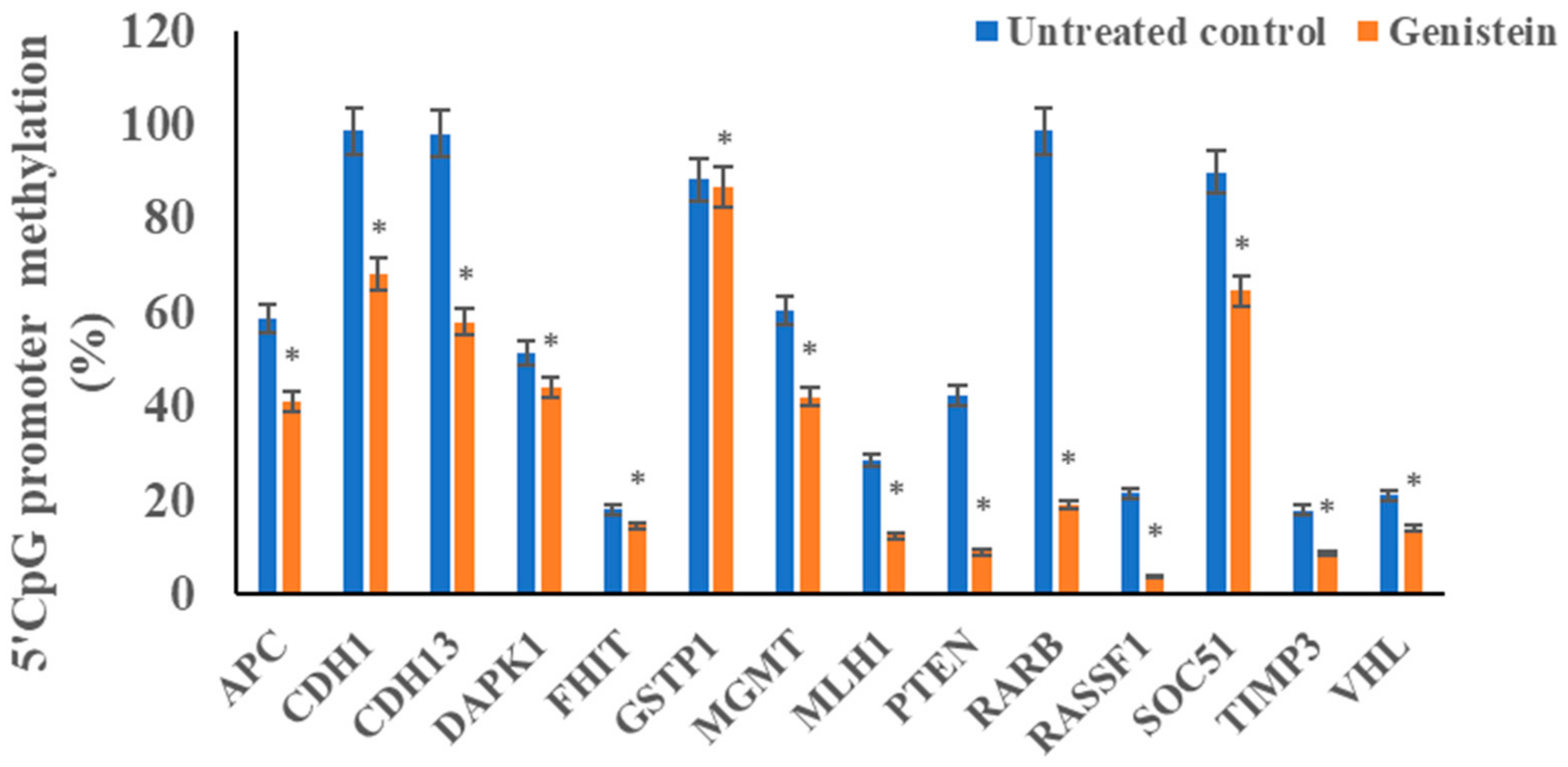
3.5. Genistein Reduces the Promoter 5’CpG Methylation of Tested Tumour Suppressor Genes
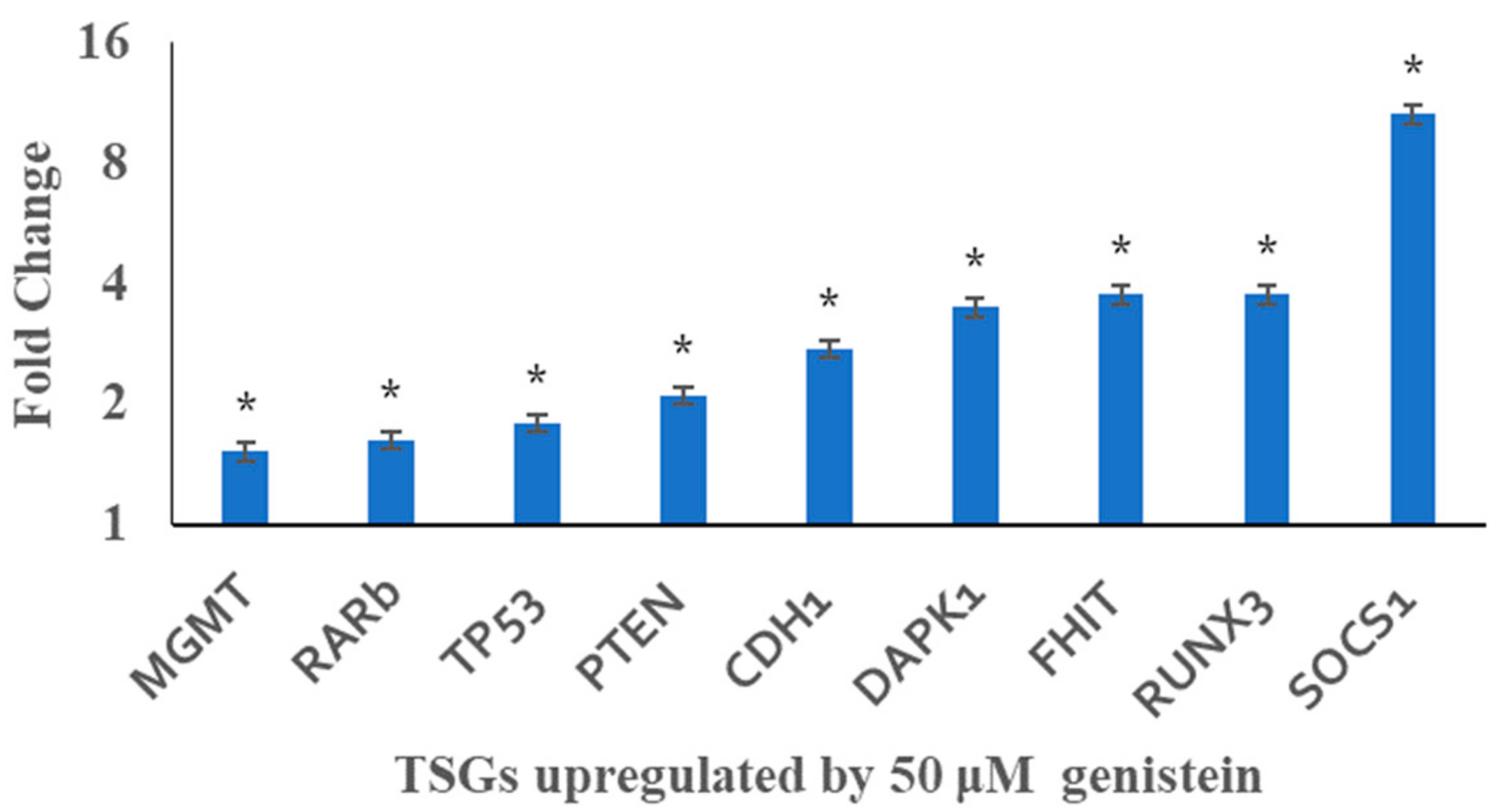
3.6. Genistein Restores the Transcription of Tested Tumour Suppressor Genes
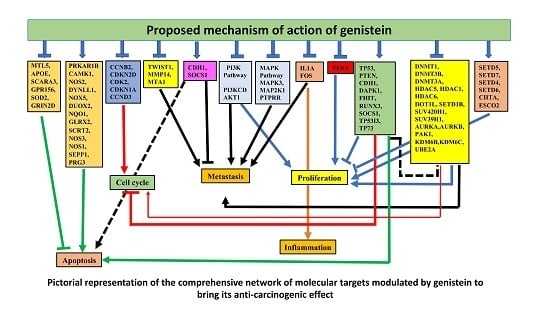
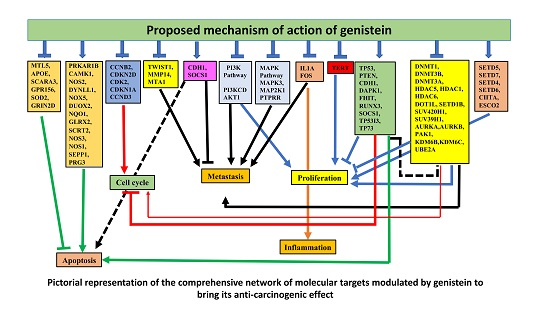
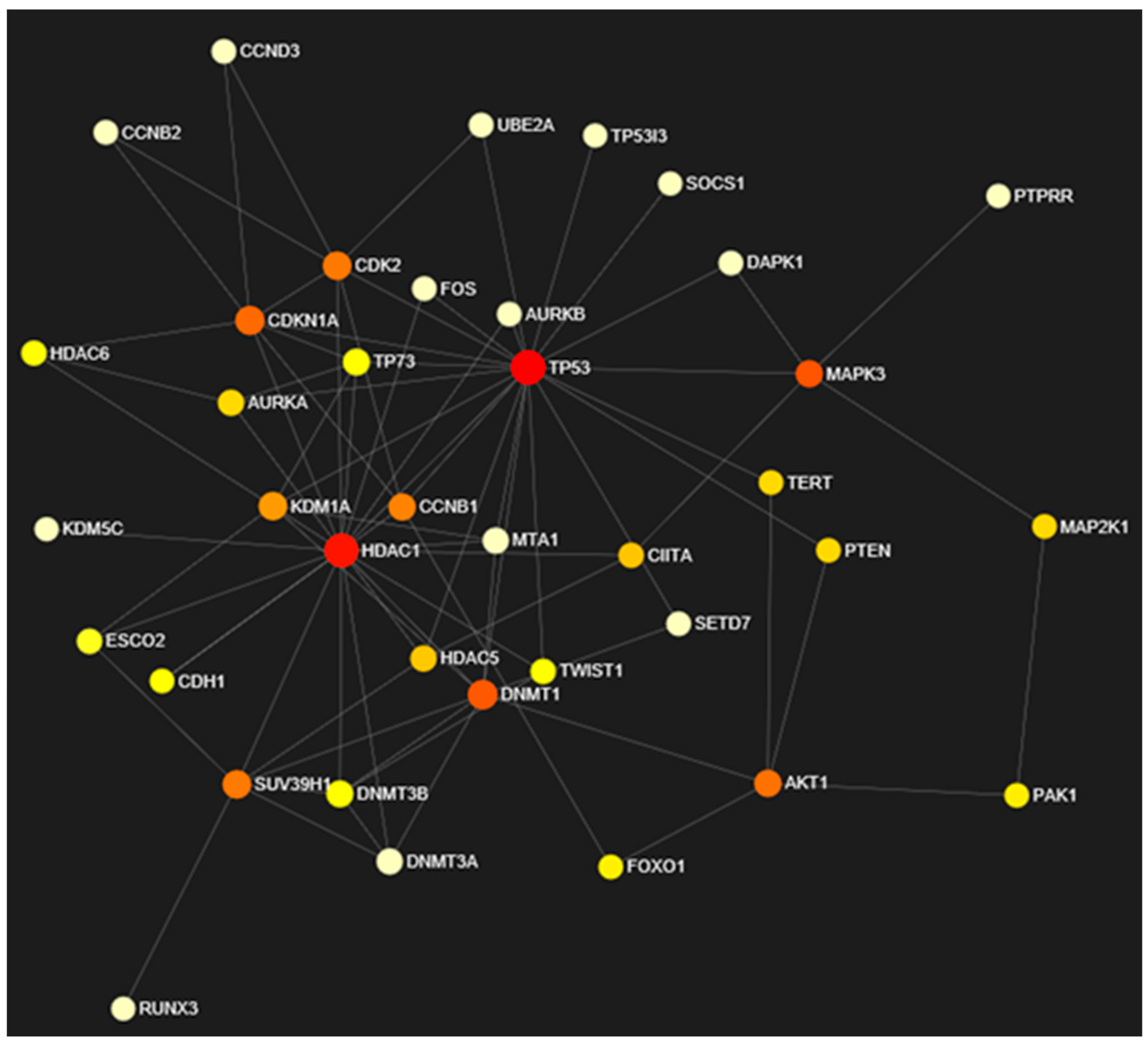
3.7. Network Analysis of Expressed Gene Sets
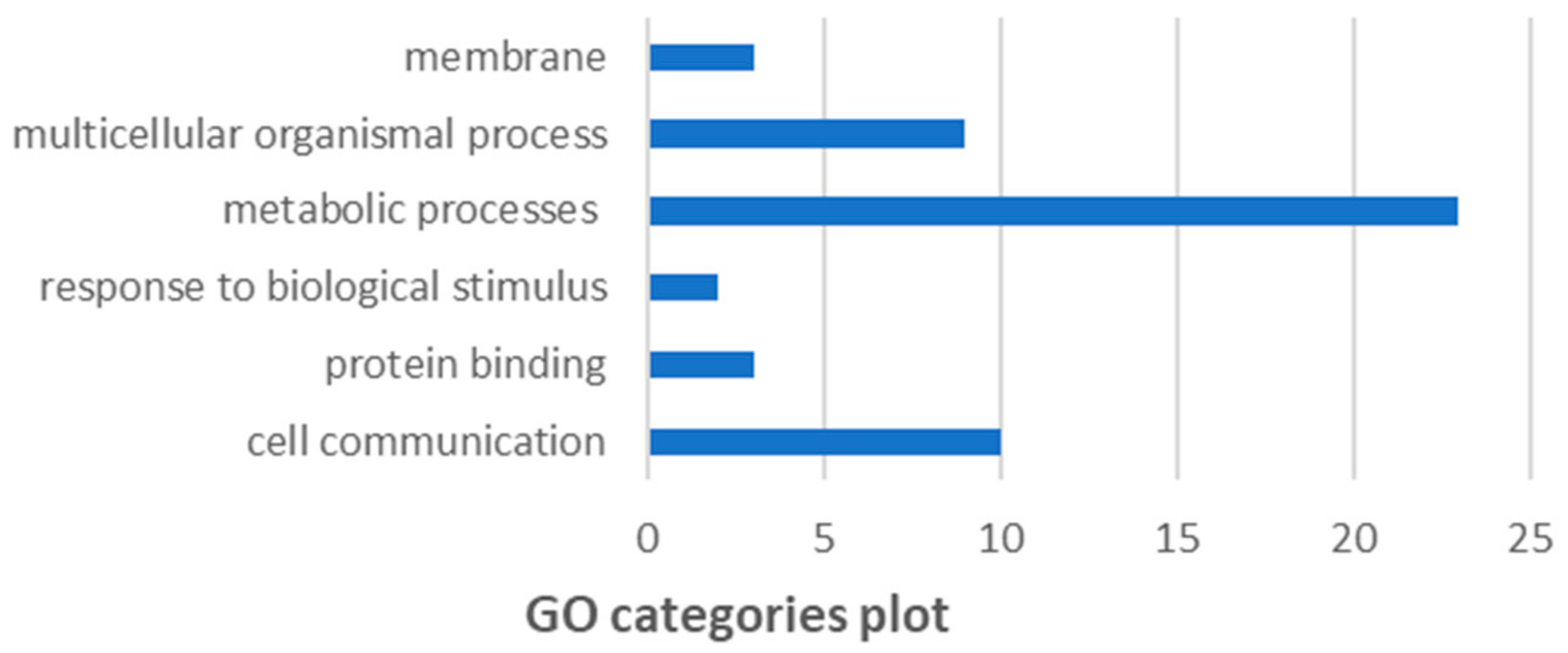
3.8. GO and KEGG pathway analysis
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Feinberg, A.P.; Tycko, B. The history of cancer epigenetics. Nat. Rev. Cancer 2004, 4, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Shankar, E.; Gupta, K.; Gupta, S. Dietary and Lifestyle Factors in Epigenetic Regulation of Cancer; Elsevier Inc.: New York, NY, USA, 2019. [Google Scholar] [CrossRef]
- Shen, H.; Laird, P.W. Interplay between the cancer genome and epigenome. Cell 2013, 153, 38–55. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Absher, D.; Brooks, J.D.; Waite, L.L.; Myers, R.M.; West, A.; Day, K.; Thalacker-Mercer, A.; Bamman, M.M. Differential DNA methylation with age displays both common and dynamic features across human tissues that are influenced by CpG landscape. Genome Biol. 2013, 14, R102. [Google Scholar] [CrossRef]
- Füllgrabe, J.; Kavanagh, E.; Joseph, B. Histone onco-modifications. Oncogene 2011, 30, 3391–3403. [Google Scholar] [CrossRef]
- Kanherkar, R.R.; Bhatia-Dey, N.; Csoka, A.B. Epigenetics across the human lifespan. Front. Cell Dev. Biol. 2014, 2, 49. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Shabashvili, D.; Li, X.; Gopalan, P.K.; Chen, M.; Zajac-kaye, M. DNA Methylation and Histone Deacetylation: Interplay and Combined Therapy in Cancer. In DNA Methylation from Genomics to Technology; Tatarinova, T., Ed.; INTECH: Rijeka, Croatia, 2011; pp. 227–288. [Google Scholar]
- Soto, D.; Song, C.; McLaughlin-Drubin, M.E. Epigenetic Alterations in Human Papillomavirus-Associated Cancers. Viruses 2017, 9, 248. [Google Scholar] [CrossRef]
- Kabekkodu, S.P.; Chakrabarty, S.; Ghosh, S.; Brand, A.; Satyamoorthy, K. Epigenomics, Pharmacoepigenomics, and Personalized Medicine in Cervical Cancer. Public Health Genomics 2017, 20, 100–115. [Google Scholar] [CrossRef]
- Mathers, J.C.; Strathdee, G.; Relton, C.L. Induction of epigenetic alterations by dietary and other environmental factors. In Advances in Genetics; Elsevier Inc.: New York, NY, USA, 2010; Volume 71, pp. 3–39. ISBN 0065-2660. [Google Scholar]
- Tao, J.J.; Visvanathan, K.; Wolff, A.C. Long term side effects of adjuvant chemotherapy in patients with early breast cancer. Breast 2016, 24, 1–12. [Google Scholar] [CrossRef]
- Van Berleere, M.; Dauchet, L. Fruits, Vegetables, and Health: Evidence From Meta-analyses of Prospective Epidemiological Studies. In Vegetarian and Plant-Based Diets in Health and Disease Prevention; Elsevier Inc.: New York, NY, USA, 2017; pp. 215–248. [Google Scholar]
- Pradhan, N.; Sengupta, D.; Patra, S.K.; Deb, M.; Parbin, S.; Das, L.; Kar, S. Epigenetic Dietary Interventions for Prevention of Cancer; Elsevier Inc.: New York, NY, USA, 2019. [Google Scholar] [CrossRef]
- Andrijauskaite, K.; Morris, J.; Wargovich, M.J. Natural Anticancer Agents; Elsevier Inc.: New York, NY, USA, 2019. [Google Scholar] [CrossRef]
- Khan, M.; Hussain, A.; Sundaram, M.; Alalami, U.; Gunasekera, D.; Ramesh, L.; Hamza, A.; Quraishi, U. (-)-Epigallocatechin-3-gallate reverses the expression of various tumor-suppressor genes by inhibiting DNA methyltransferases and histone deacetylases in human cervical cancer cells. Oncol. Rep. 2015, 1–9. [Google Scholar] [CrossRef]
- Hussain, A.; Harish, G.; Prabhu, S.A.; Mohsin, J.; Khan, M.A.; Rizvi, T. a.; Sharma, C. Inhibitory effect of genistein on the invasive potential of human cervical cancer cells via modulation of matrix metalloproteinase-9 and tissue inhibitiors of matrix metalloproteinase-1 expression. Cancer Epidemiol. 2012, 36, e387–e393. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Sramkoski, R.M.; Jacobberger, J.W. The kinetics of G2 and M transitions regulated by B cyclins. PLoS ONE 2013, 8, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Jagadeesh, S.; Banerjee, P.P. Inositol hexaphosphate represses telomerase activity and translocates TERT from the nucleus in mouse and human prostate cancer cells via the deactivation of Akt and PKCα. Biochem. Biophys. Res. Commun. 2006, 349, 1361–1367. [Google Scholar] [CrossRef] [PubMed]
- Van Doorslaer, K.; Burk, R.D. Association between hTERT activation by HPV E6 proteins and oncogenic risk. Virology 2012, 433, 216–219. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.Y.; Park, S.; Kim, S.; Lee, D.; Kim, G.; Kim, Y.; Park, K.H.; Lee, H. Use of hTERT and HPV E6/E7 mRNA RT-qPCR TaqMan assays in combination for diagnosing high-grade cervical lesions and malignant tumors. Am. J. Clin. Pathol. 2015, 143, 344–351. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R. a. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Bourboulia, D.; Stetler-Stevenson, W. Matrix MetalloProteinases (MMPs) andTissue Inhibitors of MetalloProteinases (TIMPs): positive and negative regulators intumor cell adhesion. Semin. Cancer Biol. 2011, 20, 161–168. [Google Scholar] [CrossRef]
- Scheel, C.; Weinberg, R. Cancer stem cells and epithelial–mesenchymal transition: Concepts and molecular links. Semin. Cancer Biol. 2012, 22, 396–403. [Google Scholar] [CrossRef]
- Samatov, T.R.; Tonevitsky, A.G.; Schumacher, U. Epithelial-mesenchymal transition: Focus on metastatic cascade, alternative splicing, non-coding RNAs and modulating compounds. Mol. Cancer 2013, 12. [Google Scholar] [CrossRef]
- Zhang, J.; Su, H.; Li, Q.; Li, J.; Zhao, Q. Genistein decreases A549 cell viability via inhibition of the PI3K/AKT/HIF-1α/VEGF and NF-κB/COX-2 signaling pathways. Mol. Med. Rep. 2017, 15, 2296–2302. [Google Scholar] [CrossRef]
- Su, P.H.; Lin, Y.W.; Huang, R.L.; Liao, Y.P.; Lee, H.Y.; Wang, H.C.; Chao, T.K.; Chen, C.K.; Chan, M.W.Y.; Chu, T.Y.; et al. Epigenetic silencing of PTPRR activates MAPK signaling, promotes metastasis and serves as a biomarker of invasive cervical cancer. Oncogene 2013, 32, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Candido, J.; Hagemann, T. Cancer-related inflammation. J. Clin. Immunol. 2013, 33, 79–84. [Google Scholar] [CrossRef]
- Zheng, R.; Huang, M.; Jin, C.; Wang, H.; Yu, J. Cervical cancer systemic inflammation score: a novel predictor of prognosis. Oncotarget 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Piyathilake, C.; Badiga, S.; Borak, S.; Weragoda, J.; Bae, S.; Matthews, R.; Bell, W.; Partridge, E. A higher degree of expression of DNA methyl transferase 1 in cervical cancer is associated with poor survival outcome. Int. J. Womens. Health 2017, 9, 413–420. [Google Scholar] [CrossRef]
- Sundaram, M.K.; Ansari, M.Z.; Al Mutery, A.; Ashraf, M.; Nasab, R.; Rai, S.; Rais, N.; Hussain, A. Genistein induces alterations of epigenetic modulatory signatures in human cervical cancer cells. Anticancer. Agents Med. Chem. 2017, 17, 1–11. [Google Scholar] [CrossRef]
- Feinberg, A.P. Cancer as a dysregulated epigenome allowing cellular growth advantage At the Expense of the Host. Nat. Rev. Cancer 2015, 13, 497–510. [Google Scholar]
- Kedhari Sundaram, M.; Hussain, A.; Haque, S.; Raina, R.; Afroze, N. Quercetin modifies 5′CpG promoter methylation and reactivates various tumor suppressor genes by modulating epigenetic marks in human cervical cancer cells. J. Cell. Biochem. 2019. [Google Scholar] [CrossRef]
- Kogan, A.; Unanyan, A.L.; Kadyrova, A.E.; Demura, T.A.; Sidorova, I.S.; Faizullin, R.I.; Ischenko, A.I. Immunohistochemical Analysis of Epigenetic Markers in Cervical Pathologies Associated with Human Papillomavirus Infection. Bionanoscience 2017, 7, 284–287. [Google Scholar] [CrossRef]
- Ahn, M.Y.; Yoon, J.H. Histone deacetylase 7 silencing induces apoptosis and autophagy in salivary mucoepidermoid carcinoma cells. J. Oral Pathol. Med. 2017, 46, 276–283. [Google Scholar] [CrossRef]
- Chakrabarti, A.; Oehme, I.; Witt, O.; Oliveira, G.; Sippl, W.; Romier, C.; Pierce, R.J.; Jung, M. HDAC8 : a multifaceted target for therapeutic interventions. Trends Pharmacol. Sci. 2015, 36, 481–492. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.-B.; Bin, H.; Ying-hua, P.; Shu-guang, S.; Li, Y. ESCO2 inhibits tumor metastasis via transcriptionally repressing MMP2 in colorectal cancer. Cancer Manag. Res. 2018, 10, 6157–6166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, R.-J.; Shun, C.-T.; Yen, M.-L.; Chou, C.-H.; Lin, M.-C. Methyltransferase G9a promotes cervical cancer angiogenesis and decrease patient survival. Oncotarget 2017, 8, 62081–62098. [Google Scholar]
- Wood, K.; Tellier, M.; Murphy, S. DOT1L and H3K79 methylation in transcription and genomic stability. Biomolecules 2018, 8, 11. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Liu, D.; Li, M.; Cao, C.; Wan, D.; Xi, B.; Li, W.; Tan, J.; Wang, J.; Wu, Z.; et al. Prognostic and therapeutic value of disruptor of telomeric silencing-1-like (DOT1L) expression in patients with ovarian cancer. J. Hematol. Oncol. 2017, 10, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Duan, Y.; Wu, X.; Zhao, Q.; Gao, J.; Huo, D.; Liu, X.; Ye, Z.; Dong, X.; Fu, Z.; Shang, Y.; et al. DOT1L promotes angiogenesis through cooperative regulation of VEGFR2 with ETS-1. Oncotarget 2016, 7. [Google Scholar] [CrossRef] [Green Version]
- Hsu, C.H.; Peng, K.L.; Jhang, H.C.; Lin, C.H.; Wu, S.Y.; Chiang, C.M.; Lee, S.C.; Yu, W.C.Y.; Juan, L.J. The HPV E6 oncoprotein targets histone methyltransferases for modulating specific gene transcription. Oncogene 2012, 31, 2335–2349. [Google Scholar] [CrossRef] [Green Version]
- Levy, D.; Kuo, A.J.; Chang, Y.; Schaefer, U.; Kitson, C.; Cheung, P.; Espejo, A.; Zee, B.M.; Liu, C.L.; Tangsombatvisit, S.; et al. Lysine methylation of the NF-κB subunit RelA by SETD6 couples activity of the histone methyltransferase GLP at chromatin to tonic repression of NF-κB signaling. Nat. Immunol. 2011, 12, 29–36. [Google Scholar] [CrossRef]
- Butler, J.S.; Lee, J.-H.; Skalnik, D.G. CFP1 Interacts with DNMT1 Independently of Association with the Setd1 Histone H3K4 Methyltransferase Complexes. DNA Cell Biol. 2008, 27, 533–543. [Google Scholar] [CrossRef]
- Vougiouklakis, T.; Sone, K.; Saloura, V.; Cho, H.-S.; Suzuki, T.; Dohmae, N.; Alachkar, H.; Nakamura, Y.; Hamamoto, R. SUV420H1 enhances the phosphorylation and transcription of ERK1 in cancer cells. Oncotarget 2015, 6. [Google Scholar] [CrossRef] [Green Version]
- Luo, H.; Shenoy, A.K.; Li, X.; Jin, Y.; Jin, L.; Cai, Q.; Tang, M.; Liu, Y.; Chen, H.; Reisman, D.; et al. MOF Acetylates the Histone Demethylase LSD1 to Suppress Epithelial-to-Mesenchymal Transition. Cell Rep. 2016, 15, 2665–2678. [Google Scholar] [CrossRef] [Green Version]
- Lancu Iulia, V.; Botezatu, A.; Plesa, A.; Huica, I.; Socolov, D.; Anton, G. Histone lysine demethylases as epigenetic modifiers in HPV-induced cervical neoplasia. Roman Biotechnol. Lett. 2015, 20, 10236–10244. [Google Scholar]
- Burk, R.D.; Chen, Z.; Saller, C.; Tarvin, K.; Carvalho, A.L.; Scapulatempo-Neto, C.; Silveira, H.C.; Fregnani, J.H.; Creighton, C.J.; Anderson, M.L.; et al. Integrated genomic and molecular characterization of cervical cancer. Nature 2017, 543. [Google Scholar]
- Zhang, Z.L.; Liu, G.C.; Peng, L.; Zhang, C.; Jia, Y.M.; Yang, W.H.; Mao, L. Effect of PAK1 gene silencing on proliferation and apoptosis in hepatocellular carcinoma cell lines MHCC97-H and HepG2 and cells in xenograft tumor. Gene Ther. 2018, 25, 284–296. [Google Scholar] [CrossRef]
- Xia, P.; Huang, M.; Zhang, Y.; Xiong, X.; Yan, M.; Xiong, X.; Yu, W.; Song, E. NCK1 promotes the angiogenesis of cervical squamous carcinoma via Rac1/PAK1/MMP2 signal pathway. Gynecol. Oncol. 2019, 152, 387–395. [Google Scholar] [CrossRef]
- Li, L.H.; Wu, G.Y.; Lu, Y.Z.; Chen, X.H.; Liu, B.Y.; Zheng, M.H.; Cai, J.C. P21-activated protein kinase 1 induces the invasion of gastric cancer cells through c-Jun NH2-terminal kinase-mediated activation of matrix metalloproteinase-2. Oncol. Rep. 2017, 38, 193–200. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Chang, J.-F.; Yan, H.; Wang, D.-L.; Liu, Y.; Jing, Y.; Zhang, M.; Men, Y.-L.; Lu, D.; Yang, X.-M.; et al. A conserved RAD6-MDM2 ubiquitin ligase machinery targets histone chaperone ASF1A in tumorigenesis. Oncotarget 2015, 6. [Google Scholar] [CrossRef] [Green Version]
- Shekhar, M.P.V.; Lyakhovich, A.; Visscher, D.W.; Heng, H.; Kondrat, N. Rad6 Overexpression Induces Multinucleation, Centrosome Amplification. Cancer Res. 2002, 231, 2115–2124. [Google Scholar]
- Xie, Q.; Bai, Q.; Zou, L.; Zhang, Q.; Zhou, Y.; Chang, H.; Yi, L.; Zhu, J.; Mi, M. Genistein inhibits DNA methylation and increases expression of tumor suppressor genes in human breast cancer cells. Genes Chromosom. Cancer 2014, 53, 422–431. [Google Scholar] [CrossRef]
- Bhat, S.; Kabekkodu, S.P.; Noronha, A.; Satyamoorthy, K. Biological implications and therapeutic significance of DNA methylation regulated genes in cervical cancer. Biochimie 2016, 121, 298–311. [Google Scholar] [CrossRef]
- Cardoso, M.D.F.S.; Castelletti, C.H.M.; de Lima-Filho, J.L.; Martins, D.B.G.; Teixeira, J.A.C. Putative biomarkers for cervical cancer: SNVs, methylation and expression profiles. Mutat. Res. Rev. Mutat. Res. 2017, 773, 161–173. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Li, S.; Shen, K.; Ye, S.; Cao, D.; Yang, J. DAPK1, MGMT and RARB promoter methylation as biomarkers for high-grade cervical lesions. Int. J. Clin. Exp. Pathol. 2015, 8, 14939–14945. [Google Scholar]
- Siegel, E.M.; Riggs, B.M.; Delmas, A.L.; Koch, A.; Hakam, A.; Brown, K.D. Quantitative DNA methylation analysis of candidate genes in cervical cancer. PLoS ONE 2015, 10, 1–16. [Google Scholar] [CrossRef]
- Khan, M.A.; Sundaram, M.K.; Hamza, A.; Quraishi, U.; Gunasekera, D.; Ramesh, L.; Goala, P.; Al Alami, U.; Ansari, M.Z.; Rizvi, T.A.; et al. Sulforaphane Reverses the Expression of Various Tumor Suppressor Genes by Targeting DNMT3B and HDAC1 in Human Cervical Cancer Cells. Evid. Based Complementary Altern. Med. 2015, 2015. [Google Scholar]
- Rautureau, G.J.P.; Yabal, M.; Yang, H.; Huang, D.C.S.; Kvansakul, M.; Hinds, M.G. The restricted binding repertoire of Bcl-B leaves Bim as the universal BH3-only prosurvival Bcl-2 protein antagonist. Cell Death Dis. 2012, 3, e443-9. [Google Scholar] [CrossRef]
- Ayala-Calvillo, E.; Mojica-Vazzquez, L.; Garcia-Carranca, A.; Gonzalez-Maya, L. Wnt/β-catenin pathway activation and silencing of the APC gene in HPV-positive human cervical cancer-derived cells. Mol. Med. Rep. 2017, 200–208. [Google Scholar] [CrossRef]
- Holubeková, V.; Mendelová, A.; Grendár, M.; Meršaková, S.; Kapustová, I.; Jašek, K.; Vaňochová, A.; Danko, J.; Lasabová, Z. Methylation pattern of CDH1 promoter and its association with CDH1 gene expression in cytological cervical specimens. Oncol. Lett. 2016, 12, 2613–2621. [Google Scholar] [CrossRef]
- Hao, M.; Su, X.; Zhao, W.; Wang, W.; Wang, Z.; Wang, J. Correlation between high-risk human papillomavirus expression, and death-associated protein kinase ( DAPK ) methylation status in cervical lesions. Int. J. Clin. Exp. Med. 2016, 9, 18200–18206. [Google Scholar]
- Ki, K.-D.; Lee, S.-K.; Tong, S.-Y.; Lee, J.-M.; Song, D.-H.; Chi, S.-G. Role of 5’-CpG island hypermethylation of the FHIT gene in cervical carcinoma. J. Gynecol. Oncol. 2008, 19, 117. [Google Scholar] [CrossRef]
- Banzai, C.; Nishino, K.; Quan, J.; Yoshihara, K.; Sekine, M.; Tetsuro, Y.; Tanaka, K. Promoter methylation of DAPK1, FHIT, MGMT, and CDKN2A genes in cervical carcinoma. Int. J. Clin. Oncol. 2014, 19, 127–132. [Google Scholar] [CrossRef]
- Iliopoulos, D.; Oikonomou1, P.; Messinis, I.; Tsezou, A. Correlation of promoter hypermethylation in hTERT, DAPK and and MGMT genes with cervical oncogenesis progression. Oncol. Rep. 2009, 22, 199–204. [Google Scholar]
- Ellison, A.R.; Lofing, J.; Bitter, G.A. Human MutL homolog (MLH1) function in DNA mismatch repair: a prospective screen for missense mutations in the ATPase domain. Nucleic Acids Res. 2004, 32, 5321–5338. [Google Scholar] [CrossRef] [Green Version]
- Giarnieri, E.; Mancini, R.; Pisani, T.; Alderisio, M. Msh2, Mlh1, Fhit, p53, Bcl-2, and Bax expression in invasive and in situ squamous cell carcinoma of the uterine cervix. Clin. Cancer Res. 2000, 6, 3600–3606. [Google Scholar]
- Spathis, A.; Aga, E.; Alepaki, M.; Chranioti, A.; Meristoudis, C.; Panayiotides, I.; Kassanos, D.; Karakitsos, P. Promoter methylation of p16INK4A, hMLH1, and MGMT in liquid-based cervical cytology samples compared with clinicopathological findings and HPV presence. Infect. Dis. Obstet. Gynecol. 2011, 2011. [Google Scholar] [CrossRef] [Green Version]
- Song, M.S.; Salmena, L.; Pandolfi, P.P. The functions and regulation of the PTEN tumour suppressor. Nat. Rev. Mol. Cell Biol. 2012, 13, 283. [Google Scholar] [CrossRef]
- Qi, Q.; Ling, Y.; Zhu, M.; Zhou, L.; Wan, M.; Bao, Y.; Liu, Y. Promoter region methylation and loss of protein expression of PTEN and significance in cervical cancer. Biomed. Rep. 2014, 2, 653–658. [Google Scholar] [CrossRef] [Green Version]
- Shu, R.; He, J.; Wu, C.; Gao, J. The association between RARβ and FHIT promoter methylation and the carcinogenesis of patients with cervical carcinoma: A meta-analysis. Tumor Biol. 2017, 39. [Google Scholar] [CrossRef] [Green Version]
- Donninger, H.; Vos, M.D.; Clark, G.J. The RASSF1A tumor suppressor. J. Cell Sci. 2007, 120, 3163–3172. [Google Scholar] [CrossRef] [Green Version]
- Cohen, Y.; Singer, G.; Lavie, O.; Dong, S.M.; Beller, U.; Sidransky, D. The RASSF1A tumor suppressor gene is commonly inactivated in adenocarcinoma of the uterine cervix. Clin. Cancer Res. 2003, 9. [Google Scholar]
- Narayan, G.; Arias-Pulido, H.; Koul, S.; Vargas, H.; Zhang, F.F.; Villella, J.; Schneider, A.; Terry, M.B.; Mansukhani, M.; Murty, V.V. Frequent promoter methylation of CDH1, DAPK, RARB, and HIC1 genes in carcinoma of cervix uteri: Its relationship to clinical outcome. Mol. Cancer 2003, 2, 24. [Google Scholar] [CrossRef] [Green Version]
- Deng, Y.; Nie, M.; Liu, F.; Jiang, S.; Liu, Y.; Li, D.; Meng, Q.; Li, J.; Huang, M.; Wang, M. Expression of RUNX3 in cervical carcinoma and its clinical significance. Zhong nan da xue xue bao. Yi xue ban= J. Cent. South Univ. Med. Sci. 2011, 36, 1189–1194. [Google Scholar]
- Sobti, R.C.; Singh, N.; Hussain, S.; Suri, V.; Nijhawan, R.; Bharti, A.C.; Bharadwaj, M.; Das, B.C. Aberrant promoter methylation and loss of Suppressor of Cytokine Signalling-1 gene expression in the development of uterine cervical carcinogenesis. Cell. Oncol. 2011, 34, 533–543. [Google Scholar] [CrossRef]
- Kamio, M.; Yoshida, T.; Ogata, H.; Douchi, T.; Nagata, Y.; Inoue, M.; Hasegawa, M.; Yonemitsu, Y.; Yoshimura, A. SOC1 inhibits HPV-E7-mediated transformation by inducing degradation of E7 protein. Oncogene 2004, 23, 3107–3115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henken, F.E.; Wilting, S.M.; Overmeer, R.M.; Van Rietschoten, J.G.I.; Nygren, A.O.H.; Errami, A.; Schouten, J.P.; Meijer, C.J.L.M.; Snijders, P.J.F.; Steenbergen, R.D.M. Sequential gene promoter methylation during HPV-induced cervical carcinogenesis. Br. J. Cancer 2007, 97, 1457–1464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anukriti, A.; Dhasmana, A.; Uniyal, S.; Somvanshi, P.; Bhardwaj, U.; Gupta, M.; Haque, S.; Lohani, M.; Kumar, D.; Ruokolainen, J.; et al. Investigation of precise molecular mechanistic action of tobacco associated carcinogen ‘NNK´ induced carcinogenesis: A system biology approach. Genes 2019, 10, 564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verma, A.; Somvanshi, P.; Sharda, S. Association of Inflammatory Bowel Disease with Arthritis: Evidence from In Silico Gene Expression Patterns and Network Topological Analysis. Interdiscip. Sci. Comput. Life Sci. 2017, 11, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, T.; Somvanshi, P. Pan-genome analysis of Clostridium botulinum reveals unique targets for drug development. Gene 2017, 623, 48–62. [Google Scholar] [CrossRef]
- Bhardwaj, T.; Haque, S.; Somvanshi, P. In silico identification of molecular mimics involved in the pathogenesis of Clostridium botulinum ATCC 3502 strain. Microb. Pathog. 2018, 121, 238–244. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.; Somvanshi, P.; Mishra, B.N. Reconstruction and visualization of carbohydrate, N-glycosylation pathways in Pichia pastoris CBS7435 using computational and system biology approaches. Syst. Synth. Biol. 2013, 7, 7–22. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-C.; Chan, J.Y.-H.; Chiu, Y.-L.; Liu, S.-T.; Lozano, G.; Wang, S.-L.; Ho, C.-L.; Huang, S.-M. Grail as a molecular determinant for the functions of the tumor suppressor p53 in tumorigenesis. Cell Death Differ. 2013, 20, 732–743. [Google Scholar] [CrossRef]
- Feng, D.; Wu, J.; Tian, Y.; Zhou, H.; Zhou, Y.; Hu, W.; Zhao, W.; Wei, H.; Ling, B.; Ma, C. Targeting of histone deacetylases to reactivate tumour suppressor genes and its therapeutic potential in a human cervical cancer xenograft model. PLoS ONE 2013, 8, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Song, H.; Hu, H.; Cui, L.; You, C.; Huang, L. Trichosanthin inhibits DNA methyltransferase and restores methylation-silenced gene expression in human cervical cancer cells. Mol. Med. Rep. 2012, 6, 872–878. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecular Targets | Transcriptional Expression Status | Genes Modulated by Genistein |
|---|---|---|
| Cell cycle regulation | Downregulated | CCNB1, CCNB2, CCND3, CDKN2D, CDK2, CDKN1A |
| Proliferation | Upregulated | TP53, TP73, PTEN, TP53I3 |
| Downregulated | TERT | |
| Metastasis | Upregulated | CDHI, SOCS1 |
| Downregulated | TWIST1, MMP14, MTA1 | |
| PI3K Pathway | Downregulated | PI3KCD, AKT1 |
| MAPK Pathway | Upregulated | PTPRR |
| Downregulated | MAPK3, MAP2K1 | |
| Inflammation markers | Downregulated | IL1A, FOS |
| DNA methyltransferases | Downregulated | DNMT1, DNMT3B DNMT3A |
| Histone deacetylases | Downregulated | HDAC5, HDAC1, HDAC6 |
| Histone acetylases | Upregulated | CIITA, ESCO2 |
| Downregulated | ||
| Histone methylases | Upregulated | SETD5, SETD7, SETD6 |
| Downregulated | DOT1L, SETD1B, SUV420H1, SUV39H1 | |
| Demethylases | Downregulated | KDM1A, KDM6B, KDM5C |
| Histone phosphorylases | Downregulated | AURKA, AURKB, PAK1 |
| Histone ubiquitinases | Downregulated | UBE2A |
| Tumour Suppressor Genes | Decreased promoter methylation | APC, BRCA1, CDH1, CDH13, DAPK1, FHIT, GSTP1, MGMT, MLH1, PTEN, RARB, RASSF1, SOC51, TIMP3, VHL |
| Restored expression | TP53, PTEN, CDH1, DAPK1, FHIT, RUNX3, SOCS1, TP53I3, TP73, RARβ |
| S.No. | Gene Name | Degree | Betweenness | Expression |
|---|---|---|---|---|
| 1. | TP53 | 21 | 321.87 | 1.78 |
| 2. | HDAC1 | 19 | 197.1 | 0.46 |
| 3. | DNMT1 | 9 | 70.1 | 0.31 |
| 4. | CDKN1A | 8 | 50.55 | 0.5 |
| 5. | SUV39H1 | 7 | 40.5 | 0.5 |
| 6. | CDK2 | 7 | 39.98 | 0.45 |
| 7. | KDM1A | 7 | 22.08 | 0.4 |
| 8. | MAPK3 | 5 | 74.08 | 0.4 |
| 9. | AKT1 | 5 | 44.48 | 0.45 |
| 10. | CCNB1 | 5 | 33.13 | 0.31 |
| 11. | DNMT3B | 5 | 2.2 | 0.33 |
| 12. | TP73 | 5 | 1.42 | 5.27 |
| 13. | HDAC5 | 4 | 10.24 | 0.46 |
| 14. | AURKA | 4 | 7.32 | 0.4 |
| 15. | MTA1 | 4 | 0 | 0.5 |
| 16. | DNMT3A | 4 | 0 | 0.5 |
| 17. | CIITA | 3 | 10.53 | 4.93 |
| 18. | TWIST1 | 3 | 2.95 | 0.5 |
| 19. | HDAC6 | 3 | 1.42 | 0.5 |
| 20. | ESCO2 | 3 | 1.07 | 13.93 |
| 21. | MAP2K1 | 2 | 7.39 | 0.47 |
| 22. | PTEN | 2 | 7.06 | 2.1 |
| 23. | TERT | 2 | 7.06 | 0.5 |
| 24. | PAK1 | 2 | 4.78 | 0.5 |
| 25. | FOXO1 | 2 | 4.47 | 1.89 |
| 26. | CDH1 | 2 | 2.23 | 2.76 |
| 27. | AURKB | 2 | 0 | 0.44 |
| 28. | CCNB2 | 2 | 0 | 0.5 |
| 29. | SETD7 | 2 | 0 | 2.35 |
| 30. | CCND3 | 2 | 0 | 0.5 |
| 31. | FOS | 2 | 0 | 0.37 |
| 32. | DAPK1 | 2 | 0 | 3.49 |
| 33. | UBE2A | 2 | 0 | 0.38 |
| 34. | TP53I3 | 1 | 0 | 4.96 |
| 35. | RUNX3 | 1 | 0 | 3.76 |
| 36. | SOCS1 | 1 | 0 | 10.62 |
| 37. | PTPRR | 1 | 0 | 4.03 |
| 38. | KDM5C | 1 | 0 | 0.5 |
| KEGG Pathways | Total | Expected | Hits | P.Value | FDR |
|---|---|---|---|---|---|
| p53 signaling pathway | 72 | 0.307 | 9 | 1.01e−11 | 3.21e−09 |
| Cellular senescence | 160 | 0.682 | 11 | 2.74e−11 | 4.36e−09 |
| FoxO signaling pathway | 132 | 0.563 | 10 | 9.84e−11 | 1.04e−08 |
| HTLV-I infection | 219 | 0.934 | 11 | 8.15e−10 | 6.48e−08 |
| Endometrial cancer | 58 | 0.247 | 7 | 3.36e−09 | 2.14e−07 |
| Prostate cancer | 97 | 0.414 | 8 | 4.89e−09 | 2.59e−07 |
| Pathways in cancer | 530 | 2.26 | 14 | 1.03e−08 | 4.68e−07 |
| Bladder cancer | 41 | 0.175 | 6 | 1.51e−08 | 5.59e−07 |
| Melanoma | 72 | 0.307 | 7 | 1.58e−08 | 5.59e−07 |
| Progesterone-mediated oocyte maturation | 99 | 0.422 | 7 | 1.48e−07 | 4.69e−06 |
| Thyroid cancer | 37 | 0.158 | 5 | 4.07e−07 | 1.18e−05 |
| Glioma | 75 | 0.32 | 6 | 6.1e−07 | 1.57e−05 |
| Chronic myeloid leukemia | 76 | 0.324 | 6 | 6.6e−07 | 1.57e−05 |
| Cell cycle | 124 | 0.529 | 7 | 6.93e−07 | 1.57e-05 |
| Colorectal cancer | 86 | 0.367 | 6 | 1.38e−06 | 2.85e−05 |
| Viral carcinogenesis | 201 | 0.857 | 8 | 1.43e−06 | 2.85e−05 |
| Breast cancer | 147 | 0.627 | 7 | 2.19e−06 | 4.09e−05 |
| MicroRNAs in cancer | 299 | 1.28 | 9 | 2.9e−06 | 4.98e−05 |
| Endocrine resistance | 98 | 0.418 | 6 | 2.97e−06 | 4.98e−05 |
| Hepatitis B | 163 | 0.695 | 7 | 4.36e−06 | 6.94e−05 |
| Central carbon metabolism in cancer | 65 | 0.277 | 5 | 7.09e−06 | 0.000107 |
| Non-small cell lung cancer | 66 | 0.282 | 5 | 7.64e−06 | 0.00011 |
| Thyroid hormone signaling pathway | 116 | 0.495 | 6 | 7.94e−06 | 0.00011 |
| Renal cell carcinoma | 69 | 0.294 | 5 | 9.53e−06 | 0.000126 |
| Prolactin signaling pathway | 70 | 0.299 | 5 | 1.02e−05 | 0.00013 |
| Oocyte meiosis | 125 | 0.533 | 6 | 1.22e−05 | 0.000149 |
| Pancreatic cancer | 75 | 0.32 | 5 | 1.44e−05 | 0.000169 |
| Epstein-Barr virus infection | 201 | 0.857 | 7 | 1.73e−05 | 0.00019 |
| Proteoglycans in cancer | 201 | 0.857 | 7 | 1.73e−05 | 0.00019 |
| Measles | 138 | 0.589 | 6 | 2.15e−05 | 0.000228 |
| ErbB signaling pathway | 85 | 0.363 | 5 | 2.65e−05 | 0.000272 |
| Small cell lung cancer | 93 | 0.397 | 5 | 4.1e−05 | 4e−04 |
| Hepatitis C | 155 | 0.661 | 6 | 4.15e−05 | 4e−04 |
| T cell receptor signaling pathway | 101 | 0.431 | 5 | 6.09e−05 | 0.00057 |
| PI3K-Akt signaling pathway | 354 | 1.51 | 8 | 9e−05 | 0.000818 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sundaram, M.K.; Unni, S.; Somvanshi, P.; Bhardwaj, T.; Mandal, R.K.; Hussain, A.; Haque, S. Genistein Modulates Signaling Pathways and Targets Several Epigenetic Markers in HeLa Cells. Genes 2019, 10, 955. https://doi.org/10.3390/genes10120955
Sundaram MK, Unni S, Somvanshi P, Bhardwaj T, Mandal RK, Hussain A, Haque S. Genistein Modulates Signaling Pathways and Targets Several Epigenetic Markers in HeLa Cells. Genes. 2019; 10(12):955. https://doi.org/10.3390/genes10120955
Chicago/Turabian StyleSundaram, Madhumitha Kedhari, Sreepoorna Unni, Pallavi Somvanshi, Tulika Bhardwaj, Raju K. Mandal, Arif Hussain, and Shafiul Haque. 2019. "Genistein Modulates Signaling Pathways and Targets Several Epigenetic Markers in HeLa Cells" Genes 10, no. 12: 955. https://doi.org/10.3390/genes10120955
APA StyleSundaram, M. K., Unni, S., Somvanshi, P., Bhardwaj, T., Mandal, R. K., Hussain, A., & Haque, S. (2019). Genistein Modulates Signaling Pathways and Targets Several Epigenetic Markers in HeLa Cells. Genes, 10(12), 955. https://doi.org/10.3390/genes10120955