Stability and Lability of Parental Methylation Imprints in Development and Disease



Abstract
:1. Introduction
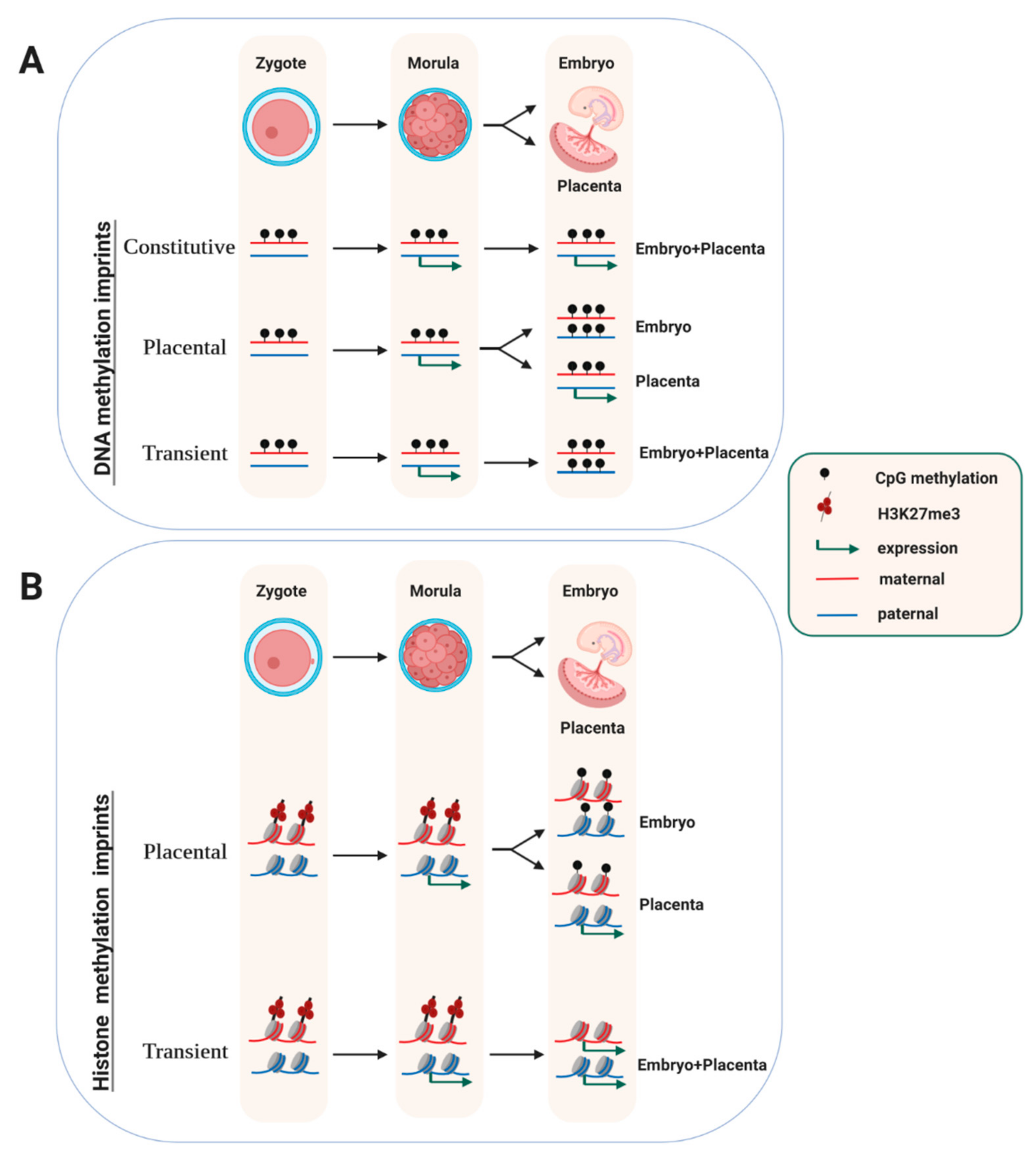
2. Embryonic Stability of Germline-Acquired DNA-Methylation Imprints
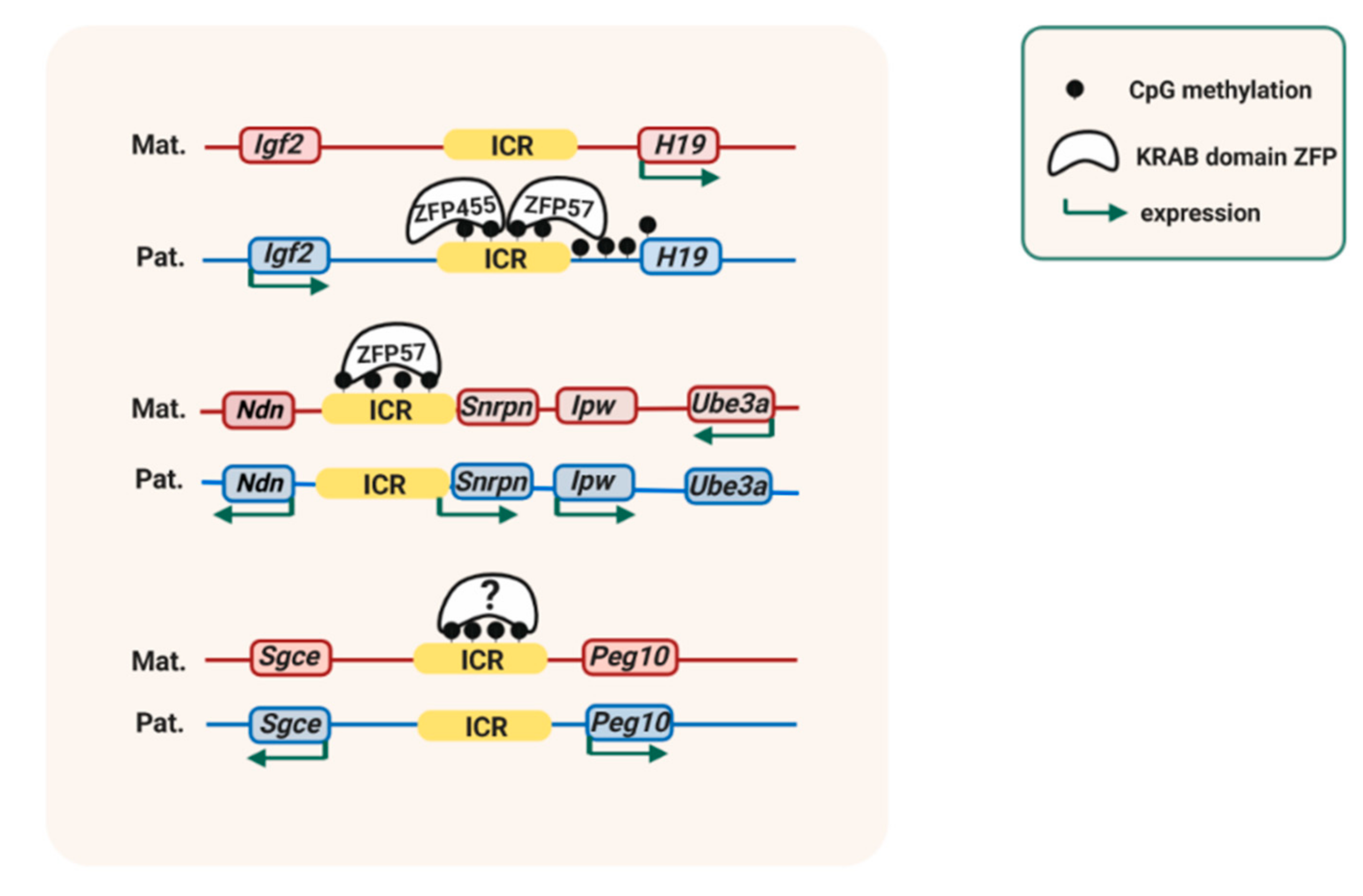
3. Transcription Factors, Histone Modifications, and Variant Histones in the Maintenance of Methylation Imprints.
4. Protection of ICRs Against de Novo DNA Methylation
5. Transient Maternally-Inherited Imprints that are Independent of DNA Methylation
6. New Insights into Maintenance Mechanisms from Imprinting Disorders
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Smith, Z.D.; Meissner, A. DNA methylation: Roles in mammalian development. Nat. Rev. Genet. 2013, 14, 204–220. [Google Scholar] [CrossRef] [PubMed]
- Iurlaro, M.; von Meyenn, F.; Reik, W. DNA methylation homeostasis in human and mouse development. Curr. Opin. Genet. Dev. 2017, 43, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Reik, W. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature 2007, 447, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Auclair, G.; Borgel, J.; Sanz, L.A.; Vallet, J.; Guibert, S.; Dumas, M.; Cavelier, P.; Girardot, M.; Forne, T.; Feil, R.; et al. Ehmt2 directs DNA methylation for efficient gene silencing in mouse embryos. Genome Res. 2016, 26, 192–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pathak, R.; Feil, R. Oocyte-derived histone h3 lysine 27 methylation controls gene expression in the early embryo. Nat. Struct. Mol. Biol. 2017, 24, 685–686. [Google Scholar] [CrossRef] [PubMed]
- Dupont, C.; Gribnau, J. Different flavors of x-chromosome inactivation in mammals. Curr. Opin. Cell Biol. 2013, 25, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef]
- Ferguson-Smith, A.C. Genomic imprinting: The emergence of an epigenetic paradigm. Nat. Rev. Genet. 2011, 12, 565–575. [Google Scholar] [CrossRef]
- Monk, D.; Mackay, D.J.G.; Eggermann, T.; Maher, E.R.; Riccio, A. Genomic imprinting disorders: Lessons on how genome, epigenome and environment interact. Nat. Rev. Genet. 2019, 20, 235–248. [Google Scholar] [CrossRef]
- Barlow, D.P. Genomic imprinting: A mammalian epigenetic discovery model. Annu. Rev. Genet. 2011, 45, 379–403. [Google Scholar] [CrossRef]
- Khamlichi, A.A.; Feil, R. Parallels between mammalian mechanisms of monoallelic gene expression. Trends Genet. 2018, 34, 954–971. [Google Scholar] [CrossRef]
- Renfree, M.B.; Suzuki, S.; Kaneko-Ishino, T. The origin and evolution of genomic imprinting and viviparity in mammals. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2013, 368, 20120151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peters, J. The role of genomic imprinting in biology and disease: An expanding view. Nat. Rev. Genet. 2014, 15, 517–530. [Google Scholar] [CrossRef]
- Plasschaert, R.N.; Bartolomei, M.S. Genomic imprinting in development, growth, behavior and stem cells. Development 2014, 141, 1805–1813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanli, I.; Feil, R. Chromatin mechanisms in the developmental control of imprinted gene expression. Int. J. Biochem. Cell Biol. 2015, 67, 139–147. [Google Scholar] [CrossRef] [Green Version]
- Bird, A.P. Cpg-rich islands and the function of DNA methylation. Nature 1986, 321, 209–213. [Google Scholar] [CrossRef]
- Kelsey, G.; Feil, R. New insights into establishment and maintenance of DNA methylation imprints in mammals. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2013, 368, 20110336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gigante, S.; Gouil, Q.; Lucattini, A.; Keniry, A.; Beck, T.; Tinning, M.; Gordon, L.; Woodruff, C.; Speed, T.P.; Blewitt, M.E.; et al. Using long-read sequencing to detect imprinted DNA methylation. Nucl. Acids Res. 2019, 47, 8. [Google Scholar] [CrossRef] [Green Version]
- Henckel, A.; Chebli, K.; Kota, S.K.; Arnaud, P.; Feil, R. Transcription and histone methylation changes correlate with imprint acquisition in male germ cells. EMBO J. 2012, 31, 606–615. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, H.; Sakurai, T.; Imai, M.; Takahashi, N.; Fukuda, A.; Yayoi, O.; Sato, S.; Nakabayashi, K.; Hata, K.; Sotomaru, Y.; et al. Contribution of intragenic DNA methylation in mouse gametic DNA methylomes to establish oocyte-specific heritable marks. PLoS Genet. 2012, 8, e1002440. [Google Scholar] [CrossRef] [Green Version]
- Smallwood, S.A.; Tomizawa, S.; Krueger, F.; Ruf, N.; Carli, N.; Segonds-Pichon, A.; Sato, S.; Hata, K.; Andrews, S.R.; Kelsey, G. Dynamic cpg island methylation landscape in oocytes and preimplantation embryos. Nat. Genet. 2011, 43, 811–814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kota, S.K.; Feil, R. Epigenetic transitions in germ cell development and meiosis. Dev. Cell 2010, 19, 675–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaneda, M.; Okano, M.; Hata, K.; Sado, T.; Tsujimoto, N.; Li, E.; Sasaki, H. Essential role for de novo DNA methyltransferase dnmt3a in paternal and maternal imprinting. Nature 2004, 429, 900–903. [Google Scholar] [CrossRef]
- Kato, Y.; Kaneda, M.; Hata, K.; Kumaki, K.; Hisano, M.; Kohara, Y.; Okano, M.; Li, E.; Nozaki, M.; Sasaki, H. Role of the dnmt3 family in de novo methylation of imprinted and repetitive sequences during male germ cell development in the mouse. Hum. Mol. Genet. 2007, 16, 2272–2280. [Google Scholar] [CrossRef]
- Bourc’his, D.; Xu, G.L.; Lin, C.S.; Bollman, B.; Bestor, T.H. Dnmt3l and the establishment of maternal genomic imprints. Science 2001, 294, 2536–2539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciccone, D.N.; Su, H.; Hevi, S.; Gay, F.; Lei, H.; Bajko, J.; Xu, G.; Li, E.; Chen, T. Kdm1b is a histone h3k4 demethylase required to establish maternal genomic imprints. Nature 2009, 461, 415–418. [Google Scholar] [CrossRef]
- Ooi, S.K.; Qiu, C.; Bernstein, E.; Li, K.; Jia, D.; Yang, Z.; Erdjument-Bromage, H.; Tempst, P.; Lin, S.P.; Allis, C.D.; et al. Dnmt3l connects unmethylated lysine 4 of histone h3 to de novo methylation of DNA. Nature 2007, 448, 714–717. [Google Scholar] [CrossRef] [Green Version]
- Stewart, K.R.; Veselovska, L.; Kim, J.; Huang, J.; Saadeh, H.; Tomizawa, S.; Smallwood, S.A.; Chen, T.; Kelsey, G. Dynamic changes in histone modifications precede de novo DNA methylation in oocytes. Genes Dev. 2015, 29, 2449–2462. [Google Scholar] [CrossRef] [Green Version]
- Dhayalan, A.; Rajavelu, A.; Rathert, P.; Tamas, R.; Jurkowska, R.Z.; Ragozin, S.; Jeltsch, A. The dnmt3a pwwp domain reads histone 3 lysine 36 trimethylation and guides DNA methylation. J. Biol. Chem. 2010, 285, 26114–26120. [Google Scholar] [CrossRef] [Green Version]
- Stewart, K.R.; Veselovska, L.; Kelsey, G. Establishment and functions of DNA methylation in the germline. Epigenomics 2016, 8, 1399–1413. [Google Scholar] [CrossRef] [Green Version]
- Smith, Z.D.; Chan, M.M.; Mikkelsen, T.S.; Gu, H.C.; Gnirke, A.; Regev, A.; Meissner, A. A unique regulatory phase of DNA methylation in the early mammalian embryo. Nature 2012, 484, U339–U374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirasawa, R.; Chiba, H.; Kaneda, M.; Tajima, S.; Li, E.; Jaenisch, R.; Sasaki, H. Maternal and zygotic dnmt1 are necessary and sufficient for the maintenance of DNA methylation imprints during preimplantation development. Genes Dev. 2008, 22, 1607–1616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pathak, R.; Feil, R. Environmental effects on chromatin repression at imprinted genes and endogenous retroviruses. Curr. Opin. Chem. Biol. 2018, 45, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.P.; Ueda, Y.; Dodge, J.E.; Wang, Z.J.; Li, E. Establishment and maintenance of genomic methylation patterns in mouse embryonic stem cells by dnmt3a and dnmt3b. Mol. Cell Biol. 2003, 23, 5594–5605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharif, J.; Muto, M.; Takebayashi, S.; Suetake, I.; Iwamatsu, A.; Endo, T.A.; Shinga, J.; Mizutani-Koseki, Y.; Toyoda, T.; Okamura, K.; et al. The sra protein np95 mediates epigenetic inheritance by recruiting dnmt1 to methylated DNA. Nature 2007, 450, 908–912. [Google Scholar] [CrossRef]
- Reese, K.J.; Lin, S.; Verona, R.I.; Schultz, R.M.; Bartolomei, M.S. Maintenance of paternal methylation and repression of the imprinted h19 gene requires mbd3. PLoS Genet. 2007, 3, e137. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Ito, M.; Zhou, F.; Youngson, N.; Zuo, X.; Leder, P.; Ferguson-Smith, A.C. A maternal-zygotic effect gene, zfp57, maintains both maternal and paternal imprints. Dev. Cell 2008, 15, 547–557. [Google Scholar] [CrossRef] [Green Version]
- Mackay, D.J.; Callaway, J.L.; Marks, S.M.; White, H.E.; Acerini, C.L.; Boonen, S.E.; Dayanikli, P.; Firth, H.V.; Goodship, J.A.; Haemers, A.P.; et al. Hypomethylation of multiple imprinted loci in individuals with transient neonatal diabetes is associated with mutations in zfp57. Nat. Genet. 2008, 40, 949–951. [Google Scholar] [CrossRef]
- Takahashi, N.; Coluccio, A.; Thorball, C.W.; Planet, E.; Shi, H.; Offner, S.; Turelli, P.; Imbeault, M.; Ferguson-Smith, A.C.; Trono, D. Znf445 is a primary regulator of genomic imprinting. Genes Dev. 2019, 33, 49–54. [Google Scholar] [CrossRef] [Green Version]
- Bohne, F.; Langer, D.; Martine, U.; Eider, C.S.; Cencic, R.; Begemann, M.; Elbracht, M.; Bulow, L.; Eggermann, T.; Zechner, U.; et al. Kaiso mediates human icr1 methylation maintenance and h19 transcriptional fine regulation. Clin. Epigenet. 2016, 8, 47. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.M.; Kim, B.J.; Norwood Toro, L.; Skoultchi, A.I. H1 linker histone promotes epigenetic silencing by regulating both DNA methylation and histone h3 methylation. Proc. Natl. Acad. Sci. USA 2013, 110, 1708–1713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girardot, M.; Hirasawa, R.; Kacem, S.; Fritsch, L.; Pontis, J.; Kota, S.K.; Filipponi, D.; Fabbrizio, E.; Sardet, C.; Lohmann, F.; et al. Prmt5-mediated histone h4 arginine-3 symmetrical dimethylation marks chromatin at g + c-rich regions of the mouse genome. Nucl. Acids Res. 2014, 42, 235–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leung, D.; Du, T.; Wagner, U.; Xie, W.; Lee, A.Y.; Goyal, P.; Li, Y.; Szulwach, K.E.; Jin, P.; Lorincz, M.C.; et al. Regulation of DNA methylation turnover at ltr retrotransposons and imprinted loci by the histone methyltransferase setdb1. Proc. Natl. Acad. Sci. USA 2014, 111, 6690–6695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, T.; Arai, Y.; Umehara, H.; Masuhara, M.; Kimura, T.; Taniguchi, H.; Sekimoto, T.; Ikawa, M.; Yoneda, Y.; Okabe, M.; et al. Pgc7/stella protects against DNA demethylation in early embryogenesis. Nat. Cell Biol. 2007, 9, 64–71. [Google Scholar] [CrossRef]
- Nakamura, T.; Liu, Y.J.; Nakashima, H.; Umehara, H.; Inoue, K.; Matoba, S.; Tachibana, M.; Ogura, A.; Shinkai, Y.; Nakano, T. Pgc7 binds histone h3k9me2 to protect against conversion of 5mc to 5hmc in early embryos. Nature 2012, 486, 415–419. [Google Scholar] [CrossRef]
- Messerschmidt, D.M.; de Vries, W.; Ito, M.; Solter, D.; Ferguson-Smith, A.; Knowles, B.B. Trim28 is required for epigenetic stability during mouse oocyte to embryo transition. Science 2012, 335, 1499–1502. [Google Scholar] [CrossRef]
- Voon, H.P.; Hughes, J.R.; Rode, C.; De La Rosa-Velazquez, I.A.; Jenuwein, T.; Feil, R.; Higgs, D.R.; Gibbons, R.J. Atrx plays a key role in maintaining silencing at interstitial heterochromatic loci and imprinted genes. Cell Rep. 2015, 11, 405–418. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.C.; Peng, S.H.; Shen, L.; Lee, C.F.; Du, T.H.; Kang, M.L.; Xu, G.L.; Upadhyay, A.K.; Cheng, X.D.; Yan, Y.T.; et al. The role of n-alpha-acetyltransferase 10 protein in DNA methylation and genomic imprinting. Mol. Cell 2017, 68, 89. [Google Scholar] [CrossRef] [Green Version]
- Meyer, E.; Lim, D.; Pasha, S.; Tee, L.J.; Rahman, F.; Yates, J.R.; Woods, C.G.; Reik, W.; Maher, E.R. Germline mutation in nlrp2 (nalp2) in a familial imprinting disorder (beckwith-wiedemann syndrome). PLoS Genet. 2009, 5, e1000423. [Google Scholar] [CrossRef] [Green Version]
- Docherty, L.E.; Rezwan, F.I.; Poole, R.L.; Turner, C.L.; Kivuva, E.; Maher, E.R.; Smithson, S.F.; Hamilton-Shield, J.P.; Patalan, M.; Gizewska, M.; et al. Mutations in nlrp5 are associated with reproductive wastage and multilocus imprinting disorders in humans. Nat. Commun. 2015, 6, 8086. [Google Scholar] [CrossRef] [Green Version]
- Ma, P.; Lin, S.; Bartolomei, M.S.; Schultz, R.M. Metastasis tumor antigen 2 (mta2) is involved in proper imprinted expression of h19 and peg3 during mouse preimplantation development. Biol. Reprod. 2010, 83, 1027–1035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, M.Y.; Tsai, T.F.; Beaudet, A.L. Deficiency of rbbp1/arid4a and rbbp1l1/arid4b alters epigenetic modifications and suppresses an imprinting defect in the pws/as domain. Genes Dev. 2006, 20, 2859–2870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gendrel, A.V.; Tang, Y.A.; Suzuki, M.; Godwin, J.; Nesterova, T.B.; Greally, J.M.; Heard, E.; Brockdorff, N. Epigenetic functions of smchd1 repress gene clusters on the inactive x chromosome and on autosomes. Mol. Cell Biol. 2013, 33, 3150–3165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Delgado, M.; Riccio, A.; Eggermann, T.; Maher, E.R.; Lapunzina, P.; MacKay, D.; Monk, D. Causes and consequences of multi-locus imprinting disturbances in humans. Trends Genet. 2016, 32, 444–455. [Google Scholar] [CrossRef] [Green Version]
- Rasmussen, K.D.; Helin, K. Role of tet enzymes in DNA methylation, development, and cancer. Genes Dev. 2016, 30, 733–750. [Google Scholar] [CrossRef]
- Guo, F.; Li, X.L.; Liang, D.; Li, T.; Zhu, P.; Guo, H.S.; Wu, X.L.; Wen, L.; Gu, T.P.; Hu, B.Q.; et al. Active and passive demethylation of male and female pronuclear DNA in the mammalian zygote. Cell Stem Cell 2014, 15, 447–458. [Google Scholar] [CrossRef] [Green Version]
- Okae, H.; Chiba, H.; Hiura, H.; Hamada, H.; Sato, A.; Utsunomiya, T.; Kikuchi, H.; Yoshida, H.; Tanaka, A.; Suyama, M.; et al. Genome-wide analysis of DNA methylation dynamics during early human development. PLoS Genet. 2014, 10, 12. [Google Scholar] [CrossRef] [Green Version]
- SanMiguel, J.M.; Bartolomei, M.S. DNA methylation dynamics of genomic imprinting in mouse development. Biol. Reprod. 2018, 99, 252–262. [Google Scholar] [CrossRef]
- Proudhon, C.; Duffie, R.; Ajjan, S.; Cowley, M.; Iranzo, J.; Carbajosa, G.; Saadeh, H.; Holland, M.L.; Oakey, R.J.; Rakyan, V.K.; et al. Protection against de novo methylation is instrumental in maintaining parent-of-origin methylation inherited from the gametes. Mol. Cell 2012, 47, 909–920. [Google Scholar] [CrossRef] [Green Version]
- Duffie, R.; Ajjan, S.; Greenberg, M.V.; Zamudio, N.; del Arenal, M.E.; Iranzo, J.; Okamoto, I.; Barbaux, S.; Fauque, P.; Bourc’his, D. The gpr1/zdbf2 locus provides new paradigms for transient and dynamic genomic imprinting in mammals. Genes Dev. 2014, 28, 463–478. [Google Scholar] [CrossRef] [Green Version]
- Greenberg, M.V.C.; Glaser, J.; Borsos, M.; El Marjou, F.; Walter, M.; Teissandier, A.; Bourc’his, D. Transient transcription in the early embryo sets an epigenetic state that programs postnatal growth. Nat. Genet. 2017, 49, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Delgado, M.; Court, F.; Vidal, E.; Medrano, J.; Monteagudo-Sanchez, A.; Martin-Trujillo, A.; Tayama, C.; Iglesias-Platas, I.; Kondova, I.; Bontrop, R.; et al. Human oocyte-derived methylation differences persist in the placenta revealing widespread transient imprinting. PLoS Genet. 2016, 12, e1006427. [Google Scholar] [CrossRef] [PubMed]
- Strogantsev, R.; Krueger, F.; Yamazawa, K.; Shi, H.; Gould, P.; Goldman-Roberts, M.; McEwen, K.; Sun, B.; Pedersen, R.; Ferguson-Smith, A.C. Allele-specific binding of zfp57 in the epigenetic regulation of imprinted and non-imprinted monoallelic expression. Genome Biol. 2015, 16, 112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanna, C.W.; Penaherrera, M.S.; Saadeh, H.; Andrews, S.; McFadden, D.E.; Kelsey, G.; Robinson, W.P. Pervasive polymorphic imprinted methylation in the human placenta. Genome Res. 2016, 26, 756–767. [Google Scholar] [CrossRef]
- Barbaux, S.; Gascoin-Lachambre, G.; Buffat, C.; Monnier, P.; Mondon, F.; Tonanny, M.B.; Pinard, A.; Auer, J.; Bessieres, B.; Barlier, A.; et al. A genome-wide approach reveals novel imprinted genes expressed in the human placenta. Epigenetics 2012, 7, 1079–1090. [Google Scholar] [CrossRef] [Green Version]
- Hamada, H.; Okae, H.; Toh, H.; Chiba, H.; Hiura, H.; Shirane, K.; Sato, T.; Suyama, M.; Yaegashi, N.; Sasaki, H.; et al. Allele-specific methylome and transcriptome analysis reveals widespread imprinting in the human placenta. Am. J. Hum. Genet. 2016, 99, 1045–1058. [Google Scholar] [CrossRef] [Green Version]
- Court, F.; Tayama, C.; Romanelli, V.; Martin-Trujillo, A.; Iglesias-Platas, I.; Okamura, K.; Sugahara, N.; Simon, C.; Moore, H.; Harness, J.V.; et al. Genome-wide parent-of-origin DNA methylation analysis reveals the intricacies of human imprinting and suggests a germline methylation-independent mechanism of establishment. Genome Res. 2014, 24, 554–569. [Google Scholar] [CrossRef] [Green Version]
- Wagschal, A.; Feil, R. Genomic imprinting in the placenta. Cytogenet. Genome Res. 2006, 113, 90–98. [Google Scholar] [CrossRef]
- Da Rocha, S.T.; Edwards, C.A.; Ito, M.; Ogata, T.; Ferguson-Smith, A.C. Genomic imprinting at the mammalian dlk1-dio3 domain. Trends Genet. 2008, 24, 306–316. [Google Scholar] [CrossRef]
- Sanli, I.; Lalevee, S.; Cammisa, M.; Perrin, A.; Rage, F.; Lleres, D.; Riccio, A.; Bertrand, E.; Feil, R. Meg3 non-coding rna expression controls imprinting by preventing transcriptional upregulation in cis. Cell Rep. 2018, 23, 337–348. [Google Scholar] [CrossRef] [Green Version]
- Ferron, S.R.; Charalambous, M.; Radford, E.; McEwen, K.; Wildner, H.; Hind, E.; Morante-Redolat, J.M.; Laborda, J.; Guillemot, F.; Bauer, S.R.; et al. Postnatal loss of dlk1 imprinting in stem cells and niche astrocytes regulates neurogenesis. Nature 2011, 475, 381–385. [Google Scholar] [CrossRef] [Green Version]
- Hutter, B.; Helms, V.; Paulsen, M. Tandem repeats in the cpg islands of imprinted genes. Genomics 2006, 88, 323–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, T.; Hara, S.; Kato, T.; Tamano, M.; Muramatsu, A.; Asahara, H.; Takada, S. A tandem repeat array in ig-dmr is essential for imprinting of paternal allele at the dlk1-dio3 domain during embryonic development. Hum. Mol. Genet. 2018, 27, 3283–3292. [Google Scholar] [CrossRef]
- Lewis, A.; Mitsuyaj, K.; Constancia, M.; Reik, W. Tandem repeat hypothesis in imprinting: Deletion of a conserved direct repeat element upstream of h19 has no effect on imprinting in the igf2-h19 region. Mol. Cell Biol. 2004, 24, 5650–5656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soares, M.L.; Edwards, C.A.; Dearden, F.L.; Ferron, S.R.; Curran, S.; Corish, J.A.; Rancourt, R.C.; Allen, S.E.; Charalambous, M.; Ferguson-Smith, M.A.; et al. Targeted deletion of a 170-kb cluster of line-1 repeats and implications for regional control. Genome Res. 2018, 28, 345–356. [Google Scholar] [CrossRef] [Green Version]
- Bian, C.J.; Yu, X.C. Pgc7 suppresses tet3 for protecting DNA methylation. Nucl. Acids Res. 2014, 42, 2893–2905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeung, W.K.A.; Brind’Amour, J.; Hatano, Y.; Yamagata, K.; Feil, R.; Lorincz, M.C.; Tachibana, M.; Shinkai, Y.; Sasaki, H. Histone h3k9 methyltransferase g9a in oocytes is essential for preimplantation development but dispensable for cg methylation protection. Cell Rep. 2019, 27, 282. [Google Scholar] [CrossRef] [Green Version]
- Wagschal, A.; Sutherland, H.G.; Woodfine, K.; Henckel, A.; Chebli, K.; Schulz, R.; Oakey, R.J.; Bickmore, W.A.; Feil, R. G9a histone methyltransferase contributes to imprinting in the mouse placenta. Mol. Cell Biol. 2008, 28, 1104–1113. [Google Scholar] [CrossRef] [Green Version]
- Ecco, G.; Imbeault, M.; Trono, D. Krab zinc finger proteins. Development 2017, 144, 2719–2729. [Google Scholar] [CrossRef] [Green Version]
- Juan, A.M.; Bartolomei, M.S. Evolving imprinting control regions: Krab zinc fingers hold the key. Genes Dev. 2019, 33, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Riso, V.; Cammisa, M.; Kukreja, H.; Anvar, Z.; Verde, G.; Sparago, A.; Acurzio, B.; Lad, S.; Lonardo, E.; Sankar, A.; et al. Zfp57 maintains the parent-of-origin-specific expression of the imprinted genes and differentially affects non-imprinted targets in mouse embryonic stem cells. Nucl. Acids Res. 2016, 44, 8165–8178. [Google Scholar] [CrossRef] [PubMed]
- Anvar, Z.; Cammisa, M.; Riso, V.; Baglivo, I.; Kukreja, H.; Sparago, A.; Girardot, M.; Lad, S.; De Feis, I.; Cerrato, F.; et al. Zfp57 recognizes multiple and closely spaced sequence motif variants to maintain repressive epigenetic marks in mouse embryonic stem cells. Nucl. Acids Res. 2016, 44, 1118–1132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pannetier, M.; Julien, E.; Schotta, G.; Tardat, M.; Sardet, C.; Jenuwein, T.; Feil, R. Pr-set7 and suv4-20h regulate h4 lysine-20 methylation at imprinting control regions in the mouse. EMBO Rep. 2008, 9, 998–1005. [Google Scholar] [CrossRef] [PubMed]
- Brustel, J.; Kirstein, N.; Izard, F.; Grimaud, C.; Prorok, P.; Cayrou, C.; Schotta, G.; Abdelsamie, A.F.; Dejardin, J.; Mechali, M.; et al. Histone h4k20 tri-methylation at late-firing origins ensures timely heterochromatin replication. EMBO J. 2017, 36, 2726–2741. [Google Scholar] [CrossRef] [PubMed]
- Kernohan, K.D.; Vernimmen, D.; Gloor, G.B.; Berube, N.G. Analysis of neonatal brain lacking atrx or mecp2 reveals changes in nucleosome density, ctcf binding and chromatin looping. Nucl. Acids Res. 2014, 42, 8356–8368. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.F.; Ou, D.S.C.; Lee, S.B.; Chang, L.H.; Lin, R.K.; Li, Y.S.; Upadhyay, A.K.; Cheng, X.D.; Wang, Y.C.; Hsu, H.S.; et al. Hnaa10p contributes to tumorigenesis by facilitating dnmt1-mediated tumor suppressor gene silencing. J. Clin. Investig. 2010, 120, 2920–2930. [Google Scholar] [CrossRef] [Green Version]
- Dorfel, M.J.; Lyon, G.J. The biological functions of naa10-from amino-terminal acetylation to human disease. Gene 2015, 567, 103–131. [Google Scholar] [CrossRef] [Green Version]
- Myklebust, L.M.; Van Damme, P.; Stove, S.I.; Dorfel, M.J.; Abboud, A.; Kalvik, T.V.; Grauffel, C.; Jonckheere, V.; Wu, Y.Y.; Swensen, J.; et al. Biochemical and cellular analysis of ogden syndrome reveals downstream nt-acetylation defects. Hum. Mol. Genet. 2015, 24, 1956–1976. [Google Scholar] [CrossRef] [Green Version]
- Singh, P.B.; Shloma, V.V.; Belyakin, S.N. Maternal regulation of chromosomal imprinting in animals. Chromosoma 2019, 128, 69–80. [Google Scholar] [CrossRef] [Green Version]
- Barlow, D.P. Methylation and imprinting: From host defense to gene regulation? Science 1993, 260, 309–310. [Google Scholar] [CrossRef]
- Cowley, M.; Oakey, R.J. Retrotransposition and genomic imprinting. Brief Funct. Genomics 2010, 9, 340–346. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Strogantsev, R.; Takahashi, N.; Kazachenka, A.; Lorincz, M.C.; Hemberger, M.; Ferguson-Smith, A.C. Zfp57 regulation of transposable elements and gene expression within and beyond imprinted domains. Epigenet. Chromatin 2019, 12, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maupetit-Mehouas, S.; Montibus, B.; Nury, D.; Tayama, C.; Wassef, M.; Kota, S.K.; Fogli, A.; Cerqueira Campos, F.; Hata, K.; Feil, R.; et al. Imprinting control regions (icrs) are marked by mono-allelic bivalent chromatin when transcriptionally inactive. Nucl. Acids Res. 2016, 44, 621–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanz, L.A.; Chamberlain, S.; Sabourin, J.C.; Henckel, A.; Magnuson, T.; Hugnot, J.P.; Feil, R.; Arnaud, P. A mono-allelic bivalent chromatin domain controls tissue-specific imprinting at grb10. EMBO J. 2008, 27, 2523–2532. [Google Scholar] [CrossRef] [Green Version]
- Fournier, C.; Goto, Y.; Ballestar, E.; Delaval, K.; Hever, A.M.; Esteller, M.; Feil, R. Allele-specific histone lysine methylation marks regulatory regions at imprinted mouse genes. EMBO J. 2002, 21, 6560–6570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernstein, B.E.; Mikkelsen, T.S.; Xie, X.; Kamal, M.; Huebert, D.J.; Cuff, J.; Fry, B.; Meissner, A.; Wernig, M.; Plath, K.; et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 2006, 125, 315–326. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.D.; Kim, H.; Ekram, M.B.; Yu, S.; Faulk, C.; Kim, J. Rex1/zfp42 as an epigenetic regulator for genomic imprinting. Hum. Mol. Genet. 2011, 20, 1353–1362. [Google Scholar] [CrossRef] [Green Version]
- Abi Habib, W.; Azzi, S.; Brioude, F.; Steunou, V.; Thibaud, N.; Das Neves, C.; Le Jule, M.; Chantot-Bastaraud, S.; Keren, B.; Lyonnet, S.; et al. Extensive investigation of the igf2/h19 imprinting control region reveals novel oct4/sox2 binding site defects associated with specific methylation patterns in beckwith-wiedemann syndrome. Hum. Mol. Genet. 2014, 23, 5763–5773. [Google Scholar] [CrossRef] [Green Version]
- Hori, N.; Yamane, M.; Kouno, K.; Sato, K. Induction of DNA demethylation depending on two sets of sox2 and adjacent oct3/4 binding sites (sox-oct motifs) within the mouse h19/insulin-like growth factor 2 (igf2) imprinted control region. J. Biol. Chem. 2012, 287, 44006–44016. [Google Scholar] [CrossRef] [Green Version]
- Franco, M.M.; Prickett, A.R.; Oakey, R.J. The role of ccctc-binding factor (ctcf) in genomic imprinting, development, and reproduction. Biol. Reprod. 2014, 91, 125. [Google Scholar] [CrossRef]
- Engel, N.; Thorvaldsen, J.L.; Bartolomei, M.S. Ctcf binding sites promote transcription initiation and prevent DNA methylation on the maternal allele at the imprinted h19/igf2 locus. Hum. Mol. Genet. 2006, 15, 2945–2954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoenherr, C.J.; Levorse, J.M.; Tilghman, S.M. Ctcf maintains differential methylation at the igf2/h19 locus. Nat. Genet. 2003, 33, 66–69. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.; Lin, C.; Woodfin, A.R.; Bartom, E.T.; Gao, X.; Smith, E.R.; Shilatifard, A. Regulation of the imprinted dlk1-dio3 locus by allele-specific enhancer activity. Genes Dev. 2016, 30, 92–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Shen, Y.; Dai, Q.; Yang, Q.; Zhang, Y.; Wang, X.; Xie, W.; Luo, Z.; Lin, C. A permissive chromatin state regulated by zfp281-aff3 in controlling the imprinted meg3 polycistron. Nucl. Acids Res. 2017, 45, 1177–1185. [Google Scholar] [CrossRef] [Green Version]
- Ishiuchi, T.; Ohishi, H.; Sato, T.; Kamimura, S.; Yorino, M.; Abe, S.; Suzuki, A.; Wakayama, T.; Suyama, M.; Sasaki, H. Zfp281 shapes the transcriptome of trophoblast stem cells and is essential for placental development. Cell Rep. 2019, 27, 1742. [Google Scholar] [CrossRef] [Green Version]
- Okashita, N.; Kumaki, Y.; Ebi, K.; Nishi, M.; Okamoto, Y.; Nakayama, M.; Hashimoto, S.; Nakamura, T.; Sugasawa, K.; Kojima, N.; et al. Prdm14 promotes active DNA demethylation through the ten-eleven translocation (tet)-mediated base excision repair pathway in embryonic stem cells. Development 2014, 141, 269–280. [Google Scholar] [CrossRef] [Green Version]
- Okashita, N.; Sakashita, N.; Ito, K.; Mitsuya, A.; Suwa, Y.; Seki, Y. Prdm14 maintains pluripotency of embryonic stem cells through tet-mediated active DNA demethylation. Biochem. Biophys. Res. Commun. 2015, 466, 138–145. [Google Scholar] [CrossRef]
- Yamaji, M.; Seki, Y.; Kurimoto, K.; Yabuta, Y.; Yuasa, M.; Shigeta, M.; Yamanaka, K.; Ohinata, Y.; Saitou, M. Critical function of prdm14 for the establishment of the germ cell lineage in mice. Nat. Genet. 2008, 40, 1016–1022. [Google Scholar] [CrossRef]
- Matsuzaki, H.; Okamura, E.; Shimotsuma, M.; Fukamizu, A.; Tanimoto, K. A randomly integrated transgenic h19 imprinting control region acquires methylation imprinting independently of its establishment in germ cells. Mol. Cell Biol. 2009, 29, 4595–4603. [Google Scholar] [CrossRef] [Green Version]
- Matsuzaki, H.; Okamura, E.; Kuramochi, D.; Ushiki, A.; Hirakawa, K.; Fukamizu, A.; Tanimoto, K. Synthetic DNA fragments bearing icr cis elements become differentially methylated and recapitulate genomic imprinting in transgenic mice. Epigenet. Chromatin 2018, 11, 36. [Google Scholar] [CrossRef]
- Puget, N.; Hirasawa, R.; Hu, N.S.; Laviolette-Malirat, N.; Feil, R.; Khamlichi, A.A. Insertion of an imprinted insulator into the igh locus reveals developmentally regulated, transcription-dependent control of v(d)j recombination. Mol. Cell Biol. 2015, 35, 529–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnaud, P.; Hata, K.; Kaneda, M.; Li, E.; Sasaki, H.; Feil, R.; Kelsey, G. Stochastic imprinting in the progeny of dnmt3l-/- females. Hum. Mol. Genet. 2006, 15, 589–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henckel, A.; Nakabayashi, K.; Sanz, L.A.; Feil, R.; Hata, K.; Arnaud, P. Histone methylation is mechanistically linked to DNA methylation at imprinting control regions in mammals. Hum. Mol. Genet. 2009, 18, 3375–3383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaufman, Y.; Heled, M.; Perk, J.; Razin, A.; Shemer, R. Protein-binding elements establish in the oocyte the primary imprint of the prader-willi/angelman syndromes domain. Proc. Natl. Acad. Sci. USA 2009, 106, 10242–10247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, A.; Jiang, L.; Lu, F.; Suzuki, T.; Zhang, Y. Maternal h3k27me3 controls DNA methylation-independent imprinting. Nature 2017, 547, 419–424. [Google Scholar] [CrossRef]
- Zheng, H.; Huang, B.; Zhang, B.J.; Xiang, Y.L.; Du, Z.H.; Xu, Q.H.; Li, Y.Y.; Wang, Q.J.; Ma, J.; Peng, X.; et al. Resetting epigenetic memory by reprogramming of histone modifications in mammals. Mol. Cell 2016, 63, 1066–1079. [Google Scholar] [CrossRef] [Green Version]
- Inoue, A.; Jiang, L.; Lu, F.; Zhang, Y. Genomic imprinting of xist by maternal h3k27me3. Genes Dev. 2017, 31, 1927–1932. [Google Scholar] [CrossRef] [Green Version]
- Hanna, C.W.; Perez-Palacios, R.; Gahurova, L.; Schubert, M.; Krueger, F.; Biggins, L.; Andrews, S.; Colome-Tatche, M.; Bourc’his, D.; Dean, W.; et al. Endogenous retroviral insertions drive non-canonical imprinting in extra-embryonic tissues. Genome Biol. 2019, 20, 225. [Google Scholar] [CrossRef]
- Zhang, W.; Chen, Z.; Yin, Q.; Zhang, D.; Racowsky, C.; Zhang, Y. Maternal-biased h3k27me3 correlates with paternal-specific gene expression in the human morula. Genes Dev. 2019, 33, 382–387. [Google Scholar] [CrossRef]
- Hanna, C.W.; Demond, H.; Kelsey, G. Epigenetic regulation in development: Is the mouse a good model for the human? Hum. Reprod. Update 2018, 24, 556–576. [Google Scholar] [CrossRef]
- Bak, M.; Boonen, S.E.; Dahl, C.; Hahnemann, J.M.D.; Mackay, D.J.D.G.; Tumer, Z.; Gronskov, K.; Temple, I.K.; Guldberg, P.; Tommerup, N. Genome-wide DNA methylation analysis of transient neonatal diabetes type 1 patients with mutations in zfp57. BMC Med. Genet. 2016, 17, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varrault, A.; Gueydan, C.; Delalbre, A.; Bellmann, A.; Houssami, S.; Aknin, C.; Severac, D.; Chotard, L.; Kahli, M.; Le Digarcher, A.; et al. Zac1 regulates an imprinted gene network critically involved in the control of embryonic growth. Dev. Cell 2006, 11, 711–722. [Google Scholar] [CrossRef] [PubMed]
- Patten, M.M.; Cowley, M.; Oakey, R.J.; Feil, R. Regulatory links between imprinted genes: Evolutionary predictions and consequences. Proc. Biol. Sci. 2016, 283, 1824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabory, A.; Ripoche, M.A.; Le Digarcher, A.; Watrin, F.; Ziyyat, A.; Forne, T.; Jammes, H.; Ainscough, J.F.; Surani, M.A.; Journot, L.; et al. H19 acts as a trans regulator of the imprinted gene network controlling growth in mice. Development 2009, 136, 3413–3421. [Google Scholar] [CrossRef] [Green Version]
- Stelzer, Y.; Sagi, I.; Yanuka, O.; Eiges, R.; Benvenisty, N. The noncoding rna ipw regulates the imprinted dlk1-dio3 locus in an induced pluripotent stem cell model of prader-willi syndrome. Nat. Genet. 2014, 46, 551–557. [Google Scholar] [CrossRef]
- Varrault, A.; Dantec, C.; Le Digarcher, A.; Chotard, L.; Bilanges, B.; Parrinello, H.; Dubois, E.; Rialle, S.; Severac, D.; Bouschet, T.; et al. Identification of plagl1/zac1 binding sites and target genes establishes its role in the regulation of extracellular matrix genes and the imprinted gene network. Nucl. Acids Res. 2017, 45, 10466–10480. [Google Scholar] [CrossRef] [Green Version]
- Begemann, M.; Rezwan, F.I.; Beygo, J.; Docherty, L.E.; Kolarova, J.; Schroeder, C.; Buiting, K.; Chokkalingam, K.; Degenhardt, F.; Wakeling, E.L.; et al. Maternal variants in nlrp and other maternal effect proteins are associated with multilocus imprinting disturbance in offspring. J. Med. Genet. 2018, 55, 497. [Google Scholar] [CrossRef] [Green Version]
- Feil, R.; Fraga, M.F. Epigenetics and the environment: Emerging patterns and implications. Nat. Rev. Genet. 2012, 13, 97–109. [Google Scholar] [CrossRef]
- Rhon-Calderon, E.A.; Vrooman, L.A.; Riesche, L.; Bartolomei, M.S. The effects of assisted reproductive technologies on genomic imprinting in the placenta. Placenta 2019, 84, 37–43. [Google Scholar] [CrossRef]
- Morita, S.; Noguchi, H.; Horii, T.; Nakabayashi, K.; Kimura, M.; Okamura, K.; Sakai, A.; Nakashima, H.; Hata, K.; Nakashima, K.; et al. Targeted DNA demethylation in vivo using dcas9-peptide repeat and scfv-tet1 catalytic domain fusions. Nat. Biotechnol. 2016, 34, 1060–1065. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Protein | Description | Methylation Phenotype due to Loss of Expression, or Knock-Down, in Somatic Cells or Embryos | References |
|---|---|---|---|
| DNA Methyltransferases (DNMTs) and Other DNA Methylation-Related Proteins: | |||
| DNMT1 | maintenance DNA methyltransferase | Loss of imprinting control region (ICR) DNA methylation. | [32] |
| DNMT3A DNMT3B | de novo DNA methyltransferases | Dnmt3a−/− embryonic stem cells (ES) cells: ICR hypomethylation Double-Knock out (KO)embryos: partial loss of ICR methylation at Rasgrf1 and Peg3. | [32,34] |
| UHRF1 (NP95) | binds hemi-methylated DNA post-replication and recruits DNMT1 | Loss of ICR methylation. | [35] |
| MBD3 | methyl CpG-binding domain protein-3 | Loss of ICR methylation at Igf2-H19 locus. | [36] |
| ZFP57 | Krüppel associated box (KRAB) domain zinc finger protein | Reduced methylation at human ICRs (PLAGL1, GRB10, and PEG3 loci). | [37,38] |
| ZFP445 | KRAB domain zinc finger protein | Human embryonic stem cells (ESCs): loss of ICR methylation at MEG3 and IGF2-H19 loci. Zfp445−/− Zfp57−/− double KO embryos: loss of methylation at almost all ICRs. | [39] |
| KAISO (ZBTB33) | Zinc Finger and Broad-complex, Tramtrack and Bric- à brac (BTB) Domain Containing-33 | Loss of ICR methylation at the human IGF2-H19 imprinted locus. | [40] |
| Histones and Other Chromatin-Related Proteins: | |||
| Histones H1 | linker histones | Triple knockout (H1c,d,e): reduced ICR methylation at Igf2-H19 and Dlk1-Dio3. | [41] |
| SETDB1 | H3 lysine-9-specific histone methyl-transferase | Loss of H3K9me3 at ICRs, loss of DNA methylation at Meg3, Nespas, Mest, Peg3. | [42,43] |
| DPPA3 (PGC7, Stella) | methylated histone (H3K9me2) binding protein | Partial loss of DNA methylation at several ICRs. | [44,45] |
| KAP1 (TRIM28) | KRAB-associated protein 1 | Partial loss of methylation at several ICRs (Igf2-H19 and Snrpn loci). | [46] |
| ATRX | H3.3 histone chaperone | Loss of histone variant H3.3 and H3K9me3 at ICRs; no reported effects on DNA methylation. | [47] |
| Other Proteins: | |||
| NAA10P (ARID1) | N-alfa-acetyltransferase 10 | Loss of ICR/DMR methylation at Trappc9-Peg13, Kcnq1-Kcnq1ot1, Mest, Snrpn, Grb10 and H19. | [48] |
| NLRP2 | cytoplasmic caterpillar family protein | Loss of methylation at ICR of KCNQ1 domain. | [49] |
| NLRP5 | cytoplasmic caterpillar family protein | Decrease in methylation PLAGL1, IGF2R, GRB10, MEST, KCNQ1OT1, MEG3 and PEG3. | [50] |
| MTA2 | Metastasis tumor antigen-2 | Partial losses of ICR methylation at Igf2-H19 and Peg3 domains. | [51] |
| RBBP1 and RBBP1L1 | Retinoblastoma (Rb)-binding proteins | Combined knockout: loss of ICR methylation at the Prader-Willi syndrome (PWS) (Snrpn) imprinted gene domain. | [52] |
| SMCHD1 | Hinge domain protein | ICR hypo-methylation at Peg12 and Snrpn. | [53] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Farhadova, S.; Gomez-Velazquez, M.; Feil, R. Stability and Lability of Parental Methylation Imprints in Development and Disease. Genes 2019, 10, 999. https://doi.org/10.3390/genes10120999
Farhadova S, Gomez-Velazquez M, Feil R. Stability and Lability of Parental Methylation Imprints in Development and Disease. Genes. 2019; 10(12):999. https://doi.org/10.3390/genes10120999
Chicago/Turabian StyleFarhadova, Sabina, Melisa Gomez-Velazquez, and Robert Feil. 2019. "Stability and Lability of Parental Methylation Imprints in Development and Disease" Genes 10, no. 12: 999. https://doi.org/10.3390/genes10120999
APA StyleFarhadova, S., Gomez-Velazquez, M., & Feil, R. (2019). Stability and Lability of Parental Methylation Imprints in Development and Disease. Genes, 10(12), 999. https://doi.org/10.3390/genes10120999