Genome Sequencing Illustrates the Genetic Basis of the Pharmacological Properties of Gloeostereum incarnatum

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Fungal Material, Sequencing, and Genome Assembly
2.2. Genome Annotation
2.3. Evolutionary Analysis and Phylogeny
2.4. Carbohydrate-Active Enzyme (CAZyme) Family Classification
2.5. Cytochrome P450 (CYP) Predictions
2.6. Secondary Metabolite Annotations
2.7. RNA Sequencing of the Two Major Developmental Stages
3. Results and Discussion
3.1. Genome Sequencing and Assembly
3.2. Gene and Repeat Sequence Prediction and Annotation
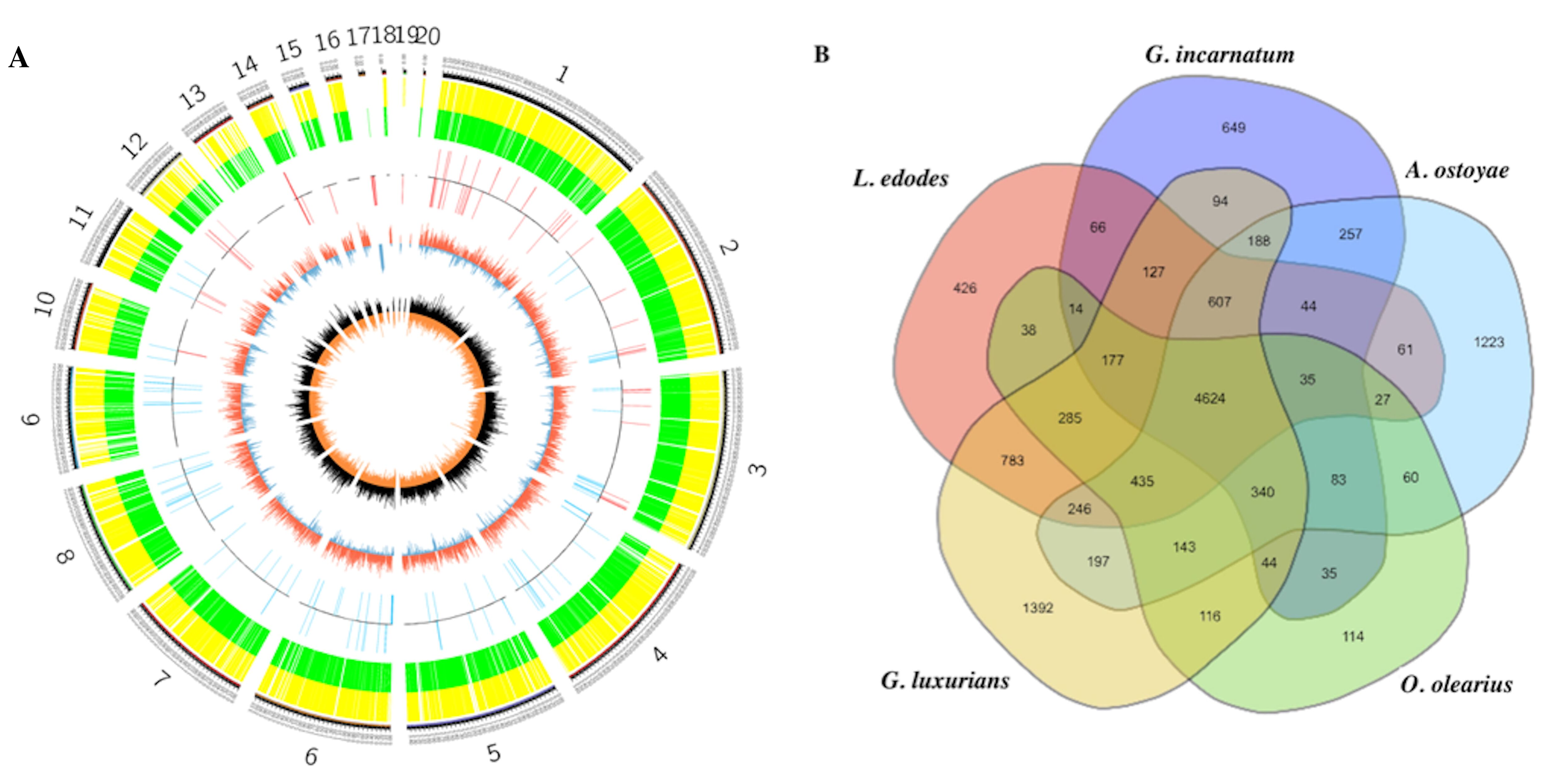
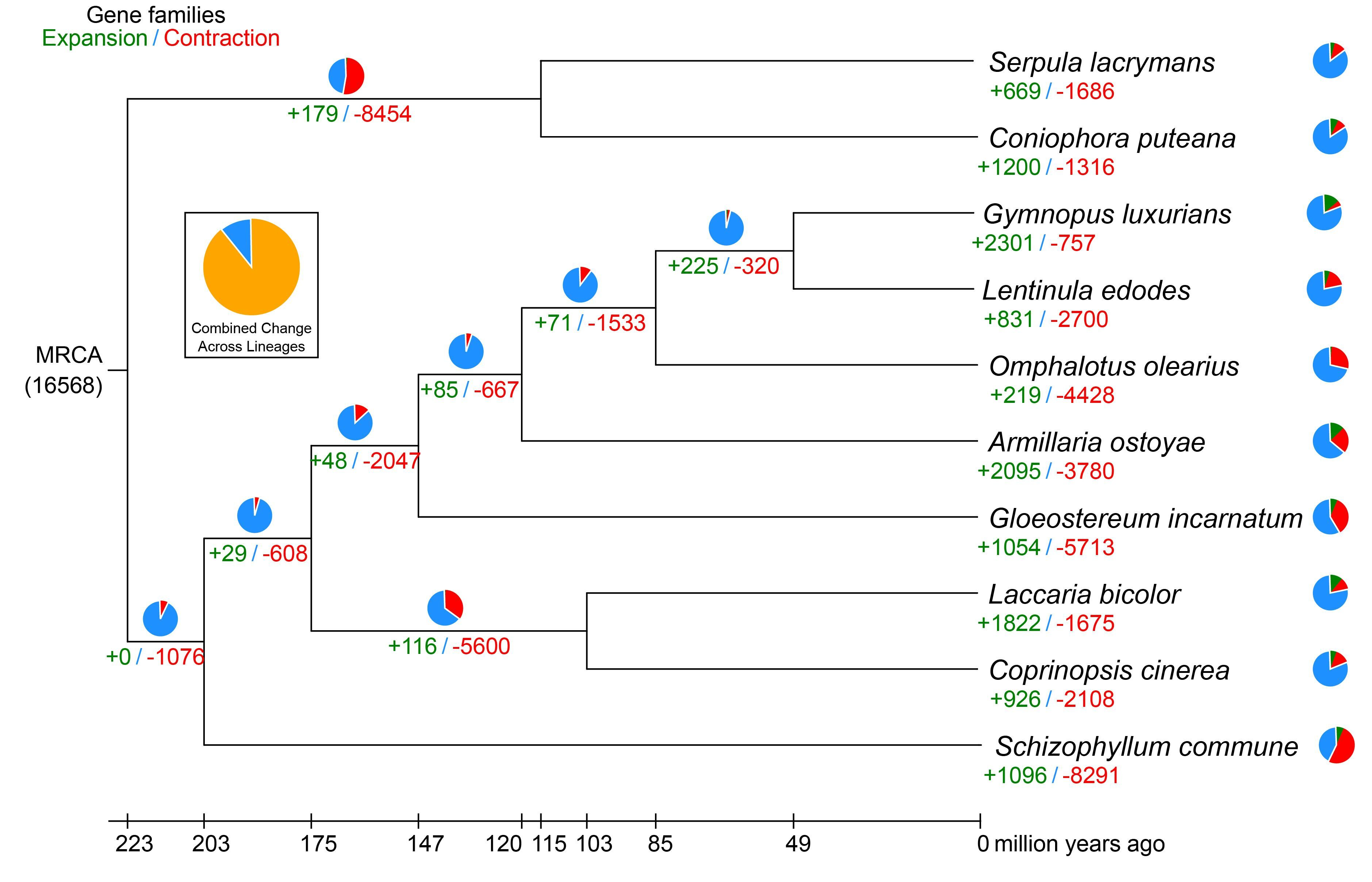
3.3. Comparative Genomics and Evolutionary Analysis
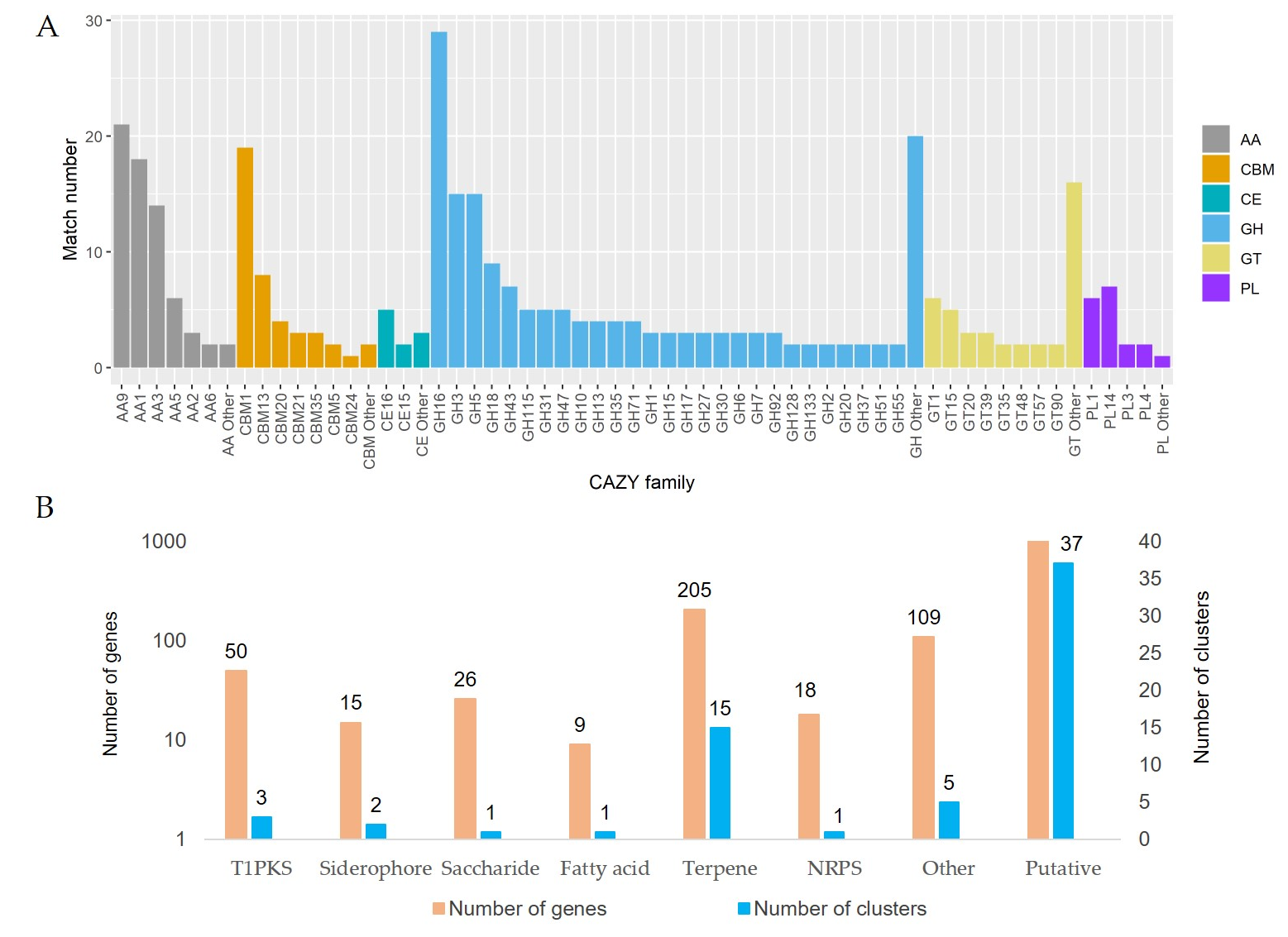
3.4. The Decomposition of Wood by CAZymes
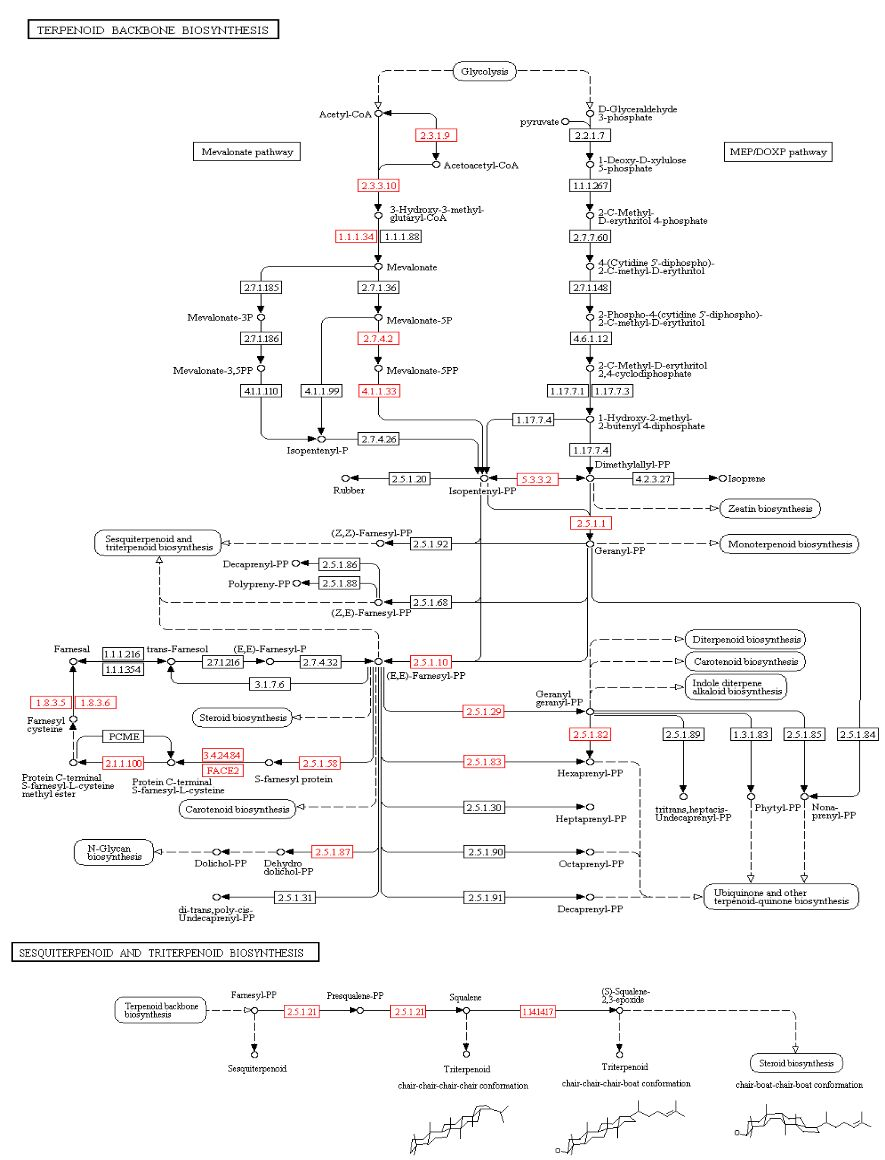
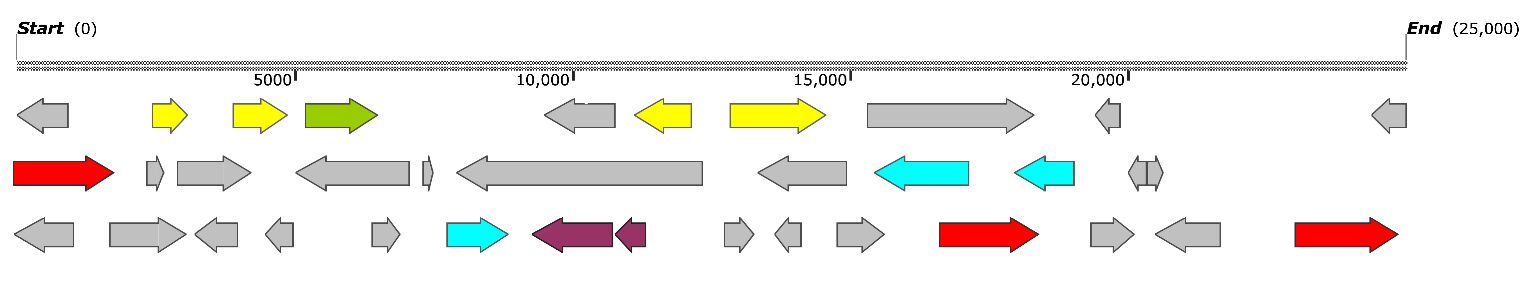
3.5. Secondary Metabolites and Terpene Pathway
3.6. The CYP Family
3.7. Polysaccharide Biosynthesis
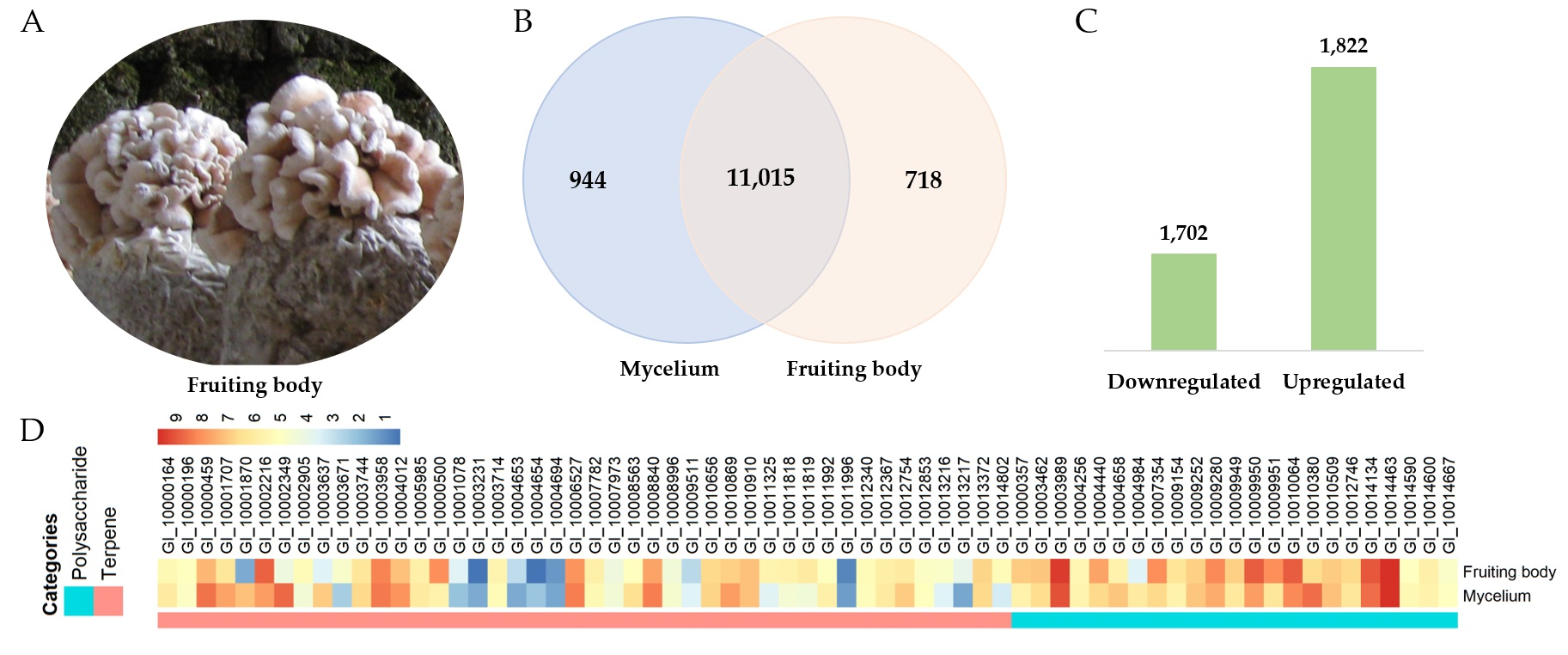
3.8. Transcriptomic Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| SMRT | Single-Molecule, Real-Time |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| CYP | cytochrome P450 |
| CAZymes | carbohydrate-active enzymes |
| DEG | differentially expressed genes |
| FPKM | fragments per kilobase of transcript per million mapped reads |
References
- Wasser, S.P. Medicinal mushroom science: Current perspectives, advances, evidences, and challenges. Biomed. J. 2014, 37, 345–356. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Liu, Y.; Song, A.; Dong, G.; Zhao, H.; Sun, W.; Ramakrishnan, S.; Wang, Y.; Wang, S.; Li, T.; et al. The Chrysanthemum nankingense genome provides insights into the evolution and diversification of chrysanthemum flowers and medicinal traits. Mol. Plant 2018, 11, 1482–1491. [Google Scholar] [CrossRef] [PubMed]
- Guggenheim, A.G.; Wright, K.M.; Zwickey, H.L. Immune modulation from five major mushrooms: Application to integrative oncology. Integr. Med. 2014, 13, 32–44. [Google Scholar]
- Petersen, R.H.; Parmasto, E. A redescription of Gloeostereum incarnatum. Mycol. Res. 1993, 97, 1213–1216. [Google Scholar] [CrossRef]
- Zhang, Z.-F.; Lv, G.-Y.; Jiang, X.; Cheng, J.-H.; Fan, L.-F. Extraction optimization and biological properties of a polysaccharide isolated from Gleoestereum incarnatum. Carbohydr. Polym. 2015, 117, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Li, Q.; Qu, Y.; Wang, M.; Li, L.; Liu, Y.; Li, Y. The investigation of immunomodulatory activities of Gloeostereum incaratum polysaccharides in cyclophosphamide-induced immunosuppression mice. Exp. Ther. Med. 2018, 15, 3633–3638. [Google Scholar] [CrossRef] [PubMed]
- Lull, C.; Wichers, H.J.; Savelkoul, H.F. Antiinflammatory and immunomodulating properties of fungal metabolites. Mediat. Inflamm. 2005, 2005, 63–80. [Google Scholar] [CrossRef] [PubMed]
- Asai, R.; Mitsuhashi, S.; Shigetomi, K.; Miyamoto, T.; Ubukata, M. Absolute configurations of (−)-hirsutanol A and (−)-hirsutanol C produced by Gloeostereum incarnatum. J. Antibiot. 2011, 64, 693–696. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Chen, L.; Cai, Y.; Zhang, Q.; Bian, Y. Opposite polarity monospore genome de novo sequencing and comparative analysis rreveal the possible heterothallic life cycle of Morchella importuna. Int. J. Mol. Sci. 2018, 19, 2525. [Google Scholar] [CrossRef]
- Dai, Y.; Su, W.; Yang, C.; Song, B.; Li, Y.; Fu, Y. Development of novel polymorphic EST-SSR markers in Bailinggu (Pleurotus tuoliensis) for crossbreeding. Genes 2017, 8, 325. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.Y.; Fan, W.L.; Wang, W.F.; Chen, T.; Tang, Y.C.; Chu, F.H.; Chang, T.T.; Wang, S.Y.; Li, M.Y.; Chen, Y.H.; et al. Genomic and transcriptomic analyses of the medicinal fungus Antrodia cinnamomea for its metabolite biosynthesis and sexual development. Proc. Natl. Acad. Sci. USA 2014, 111, E4743–E4752. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Xu, J.; Liu, C.; Zhu, Y.; Nelson, D.R.; Zhou, S.; Li, C.; Wang, L.; Guo, X.; Sun, Y.; et al. Genome sequence of the model medicinal mushroom Ganoderma lucidum. Nat. Commun. 2012, 3, 913. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zeng, X.; Yang, Y.L.; Xing, Y.M.; Zhang, Q.; Li, J.M.; Ma, K.; Liu, H.W.; Guo, S.X. Genomic and transcriptomic analyses reveal differential regulation of diverse terpenoid and polyketides secondary metabolites in Hericium erinaceus. Sci. Rep. 2017, 7, 10151. [Google Scholar] [CrossRef] [PubMed]
- Takazawa, H.; Kashino, S. Incarnal. A new antibacterial sesquiterpene from Basidiomycetes. Chem. Pharm. Bull. 1991, 39, 555–557. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Lin, F.; An, D.; Wang, W.; Huang, R. Genome sequencing and assembly by long reads in plants. Genes 2017, 9, 6. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Dai, Y.; Yang, C.; Wei, P.; Song, B.; Yang, Y.; Sun, L.; Zhang, Z.W.; Li, Y. Comparative transcriptome analysis identified candidate genes related to Bailinggu mushroom formation and genetic markers for genetic analyses and breeding. Sci. Rep. 2017, 7, 9266. [Google Scholar] [CrossRef] [PubMed]
- Parra, G.; Bradnam, K.; Korf, I. CEGMA: A pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 2007, 23, 1061–1067. [Google Scholar] [CrossRef] [PubMed]
- Simao, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed]
- Stanke, M.; Keller, O.; Gunduz, I.; Hayes, A.; Waack, S.; Morgenstern, B. AUGUSTUS: Ab initio prediction of alternative transcripts. Nucleic Acids Res. 2006, 34, W435–W439. [Google Scholar] [CrossRef] [PubMed]
- Burge, C.; Karlin, S. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 1997, 268, 78–94. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.E.; Majoros, W.H.; Pertea, M.; Salzberg, S.L. JIGSAW, GeneZilla, and GlimmerHMM: Puzzling out the features of human genes in the ENCODE regions. Genome Biol. 2006, 7 (Suppl. 1), S9. [Google Scholar] [CrossRef]
- Korf, I. Gene finding in novel genomes. BMC Bioinform. 2004, 5, 59. [Google Scholar] [CrossRef] [PubMed]
- Elsik, C.G.; Mackey, A.J.; Reese, J.T.; Milshina, N.V.; Roos, D.S.; Weinstock, G.M. Creating a honey bee consensus gene set. Genome Biol. 2007, 8, R13. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The gene ontology consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A.; Koonin, E.V. The COG database: A tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef] [PubMed]
- Jurka, J.; Kapitonov, V.V.; Pavlicek, A.; Klonowski, P.; Kohany, O.; Walichiewicz, J. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 2005, 110, 462–467. [Google Scholar] [CrossRef] [PubMed]
- Tarailo-Graovac, M.; Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinform. 2009. [Google Scholar] [CrossRef]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Lagesen, K.; Hallin, P.; Rodland, E.A.; Staerfeldt, H.H.; Rognes, T.; Ussery, D.W. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007, 35, 3100–3108. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Gardner, P.P.; Daub, J.; Tate, J.G.; Nawrocki, E.P.; Kolbe, D.L.; Lindgreen, S.; Wilkinson, A.C.; Finn, R.D.; Griffiths-Jones, S.; Eddy, S.R.; et al. Rfam: Updates to the RNA families database. Nucleic Acids Res. 2009, 37, D136–D140. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed]
- Cantarel, B.L.; Coutinho, P.M.; Rancurel, C.; Bernard, T.; Lombard, V.; Henrissat, B. The Carbohydrate-Active EnZymes database (CAZy): An expert resource for Glycogenomics. Nucleic Acids Res. 2009, 37, D233–D238. [Google Scholar] [CrossRef] [PubMed]
- Nelson, D.R. The cytochrome p450 homepage. Hum. Genom. 2009, 4, 59–65. [Google Scholar]
- Weber, T.; Blin, K.; Duddela, S.; Krug, D.; Kim, H.U.; Bruccoleri, R.; Lee, S.Y.; Fischbach, M.A.; Muller, R.; Wohlleben, W.; et al. antiSMASH 3.0—A comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acids Res. 2015, 43, W237–W243. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed]
- Shim, D.; Park, S.G.; Kim, K.; Bae, W.; Lee, G.W.; Ha, B.S.; Ro, H.S.; Kim, M.; Ryoo, R.; Rhee, S.K.; et al. Whole genome de novo sequencing and genome annotation of the world popular cultivated edible mushroom, Lentinula edodes. J. Biotechnol. 2016, 223, 24–25. [Google Scholar] [CrossRef] [PubMed]
- Gupta, D.K.; Ruhl, M.; Mishra, B.; Kleofas, V.; Hofrichter, M.; Herzog, R.; Pecyna, M.J.; Sharma, R.; Kellner, H.; Hennicke, F.; et al. The genome sequence of the commercially cultivated mushroom Agrocybe aegerita reveals a conserved repertoire of fruiting-related genes and a versatile suite of biopolymer-degrading enzymes. BMC Genom. 2018, 19, 48. [Google Scholar] [CrossRef] [PubMed]
- Shu, S.; Chen, B.; Zhou, M.; Zhao, X.; Xia, H.; Wang, M. De novo sequencing and transcriptome analysis of Wolfiporia cocos to reveal genes related to biosynthesis of triterpenoids. PLoS ONE 2013, 8, e71350. [Google Scholar] [CrossRef] [PubMed]
- Morin, E.; Kohler, A.; Baker, A.R.; Foulongne-Oriol, M.; Lombard, V.; Nagye, L.G.; Ohm, R.A.; Patyshakuliyeva, A.; Brun, A.; Aerts, A.L.; et al. Genome sequence of the button mushroom Agaricus bisporus reveals mechanisms governing adaptation to a humic-rich ecological niche. Proc. Natl. Acad. Sci. USA 2012, 109, 17501–17506. [Google Scholar] [CrossRef] [PubMed]
- Yap, H.Y.; Chooi, Y.H.; Firdaus-Raih, M.; Fung, S.Y.; Ng, S.T.; Tan, C.S.; Tan, N.H. The genome of the Tiger Milk mushroom, Lignosus rhinocerotis, provides insights into the genetic basis of its medicinal properties. BMC Genom. 2014, 15, 635. [Google Scholar] [CrossRef] [PubMed]
- Xiao, D.; Ma, L.; Yang, C.; Ying, Z.; Jiang, X.; Lin, Y.Q. De novo sequencing of a Sparassis latifolia genome and its associated comparative analyses. Can. J. Infect. Dis. Med. Microbiol. 2018, 2018, 1857170. [Google Scholar] [CrossRef] [PubMed]
- Kurata, A.; Fukuta, Y.; Mori, M.; Kishimoto, N.; Shirasaka, N. Draft genome sequence of the basidiomycetous fungus Flammulina velutipes TR19. Genome Announc. 2016, 4. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.J.; Xiao, J.H. Secondary metabolites from higher fungi: Discovery, bioactivity, and bioproduction. Adv. Biochem. Eng. Biotechnol. 2009, 113, 79–150. [Google Scholar] [CrossRef] [PubMed]
- Floudas, D.; Binder, M.; Riley, R.; Barry, K.; Blanchette, R.A.; Henrissat, B.; Martinez, A.T.; Otillar, R.; Spatafora, J.W.; Yadav, J.S.; et al. The Paleozoic origin of enzymatic lignin decomposition reconstructed from 31 fungal genomes. Science 2012, 336, 1715–1719. [Google Scholar] [CrossRef] [PubMed]
- Sista Kameshwar, A.K.; Qin, W. Comparative study of genome-wide plant biomass-degrading CAZymes in white rot, brown rot and soft rot fungi. Mycology 2018, 9, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Martinez, A.T.; Ruiz-Duenas, F.J.; Martinez, M.J.; Del Rio, J.C.; Gutierrez, A. Enzymatic delignification of plant cell wall: From nature to mill. Curr. Opin. Biotechnol. 2009, 20, 348–357. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.Y.; Moon, S.; Shim, D.; Hong, C.P.; Lee, Y.; Koo, C.D.; Chung, J.W.; Ryu, H. Development of 44 novel polymorphic SSR markers for determination of shiitake mushroom (Lentinula edodes) cultivars. Genes 2017, 8, 109. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Yue, D.C.; Cheng, K.D.; Wang, S.C.; Yu, K.B.; Zheng, Q.T.; Yang, J.S. Gloeosteretriol, a new sesquiterpene from the fermentation products of Gloeostereum incarnatum S. Ito et Imai. Yao Xue Xue Bao = Acta Pharm. Sin. 1992, 27, 33–36. [Google Scholar]
- Li, H.J.; Chen, T.; Xie, Y.L.; Chen, W.D.; Zhu, X.F.; Lan, W.J. Isolation and structural elucidation of chondrosterins F-H from the marine fungus Chondrostereum sp. Mar. Drugs 2013, 11, 551–558. [Google Scholar] [CrossRef] [PubMed]
- Christianson, D.W. Unearthing the roots of the terpenome. Curr. Opin. Chem. Biol. 2008, 12, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, P.I.; Ullan, R.V.; Albillos, S.M.; Montero, O.; Fernandez-Bodega, M.A.; Garcia-Estrada, C.; Fernandez-Aguado, M.; Martin, J.F. Molecular characterization of the PR-toxin gene cluster in Penicillium roqueforti and Penicillium chrysogenum: Cross talk of secondary metabolite pathways. Fungal Genet. Biol. FG B 2014, 62, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Schothorst, R.C.; van Egmond, H.P. Report from SCOOP task 3.2.10 “collection of occurrence data of Fusarium toxins in food and assessment of dietary intake by the population of EU member states”. Subtask: Trichothecenes. Toxicol. Lett. 2004, 153, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Benveniste, P. Biosynthesis and accumulation of sterols. Annu. Rev. Plant Biol. 2004, 55, 429–457. [Google Scholar] [CrossRef] [PubMed]
- Rios, J.L.; Andujar, I.; Recio, M.C.; Giner, R.M. Lanostanoids from fungi: A group of potential anticancer compounds. J. Nat. Prod. 2012, 75, 2016–2044. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Dannert, C. Biosynthesis of terpenoid natural products in fungi. Adv. Biochem. Eng. Biotechnol. 2015, 148, 19–61. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Cresnar, B.; Petric, S. Cytochrome P450 enzymes in the fungal kingdom. Biochim. Biophys. Acta 2011, 1814, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Sanglard, D.; Loper, J.C. Characterization of the alkane-inducible cytochrome P450 (P450alk) gene from the yeast Candida tropicalis: Identification of a new P450 gene family. Gene 1989, 76, 121–136. [Google Scholar] [CrossRef]
- Mansuy, D. The great diversity of reactions catalyzed by cytochromes P450. Comp. Biochem. Physiol. Part C Pharmacol. Toxicol. Endocrinol. 1998, 121, 5–14. [Google Scholar] [CrossRef]
- Park, J.; Lee, S.; Choi, J.; Ahn, K.; Park, B.; Park, J.; Kang, S.; Lee, Y.H. Fungal cytochrome P450 database. BMC Genom. 2008, 9, 402. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Chen, X.; Zhong, Z.; Chen, L.; Wang, Y. Ganoderma lucidum polysaccharides: Immunomodulation and potential anti-tumor activities. Am. J. Chin. Med. 2011, 39, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Montijn, R.C.; Vink, E.; Muller, W.H.; Verkleij, A.J.; Van Den Ende, H.; Henrissat, B.; Klis, F.M. Localization of synthesis of beta1,6-glucan in Saccharomyces cerevisiae. J. Bacteriol. 1999, 181, 7414–7420. [Google Scholar] [PubMed]
- Yuan, Y.; Wu, F.; Si, J.; Zhao, Y.F.; Dai, Y.C. Whole genome sequence of Auricularia heimuer (Basidiomycota, Fungi), the third most important cultivated mushroom worldwide. Genomics 2017. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Organism | Accession | Genome Size (Mbp) | Genome | Scaffold | N50 (Kbp) | GC Content (%) | Protein-Coding Genes | Sequencing Method |
|---|---|---|---|---|---|---|---|---|
| Gloeostereum incarnatum | 38.7 | 94× | 20 | 3500 | 49.0 | 15,251 | PacBio Sequel | |
| Lentinula edodes | LSDU00000000 | 46.1 | 60× | 31 | 3663 | 45.3 | 13,426 | PacBio RSII; Illumina HiSeq 2500 |
| Agrocybe aegerita | PRJEB21917 | 44.8 | 253× | 122 | 768 | 49.2 | 14,113 | PacBio RSII; Illumina HiSeq 2500 |
| Hericium erinaceus | PRJN361338 | 39.4 | 200× | 519 | 538 | 53.1 | 9895 | Illumina MiSeq; Hiseq 2500 |
| Antrodia cinnamomea | JNBV00000000 | 32.2 | 878× | 360 | 1035 | 50.6 | 9254 | Roche 454; Illumina GAIIx |
| Ganoderma lucidum | AGAX00000000 | 43.3 | 440× | 82 | 1388 | 55.9 | 16,113 | Roche 454; Illumina GAII |
| Wolfiporia cocos | AEHD00000000 | 50.5 | 40× | 348 | 2539 | 52.2 | 12,212 | Sanger; Roche 454 |
| Inonotus baumii | LNZH00000000 | 31.6 | 186× | 217 | 267 | 47.6 | 8455 | Illumina HiSeq |
| Agaricus bisporus var. bisporus | AEOK00000000 | 30.2 | 8.3× | 29 | 2300 | 46.6 | 10,438 | Sanger |
| Lignosus rhinocerotis | AXZM00000000 | 34.3 | 180× | 1338 | 90 | 53.7 | 10,742 | Illumina Hiseq 2000 |
| Sparassis latifolia | LWKX00000000 | 48.1 | 601× | 472 | 641 | 51.4 | 12,471 | Illumina HiSeq 2500 |
| Flammulina velutipes | BDAN00000000 | 35.3 | 132× | 5130 | 150 | 49.6 | 13,843 | Illumina HiSeq 2500 |
| Family | Subfamily | Corresponding Gene Number | Total Gene Number | Family | Subfamily | Corresponding Gene Number | Total Gene Number |
|---|---|---|---|---|---|---|---|
| CYP5144 | C,F | 15,1 | 16 | CYP675 | A | 3 | 3 |
| CYP620 | A,B,E,H | 1,1,4,2 | 8 | CYP682 | B | 3 | 3 |
| CYP5015 | C | 6 | 6 | CYP504 | A | 3 | 3 |
| CYP5014 | F,H | 2,3 | 5 | CYP51 | F | 3 | 3 |
| CYP5068 | B | 5 | 5 | CYP55 | A | 3 | 3 |
| CYP5080 | B,D | 3,2 | 5 | CYP65 | J,X | 1,1 | 2 |
| CYP5093 | A | 5 | 5 | CYP5070 | A | 2 | 2 |
| CYP505 | C,D | 3,1 | 4 | CYP5074 | A | 2 | 2 |
| CYP535 | A | 4 | 4 | CYP5078 | A | 2 | 2 |
| CYP536 | A | 4 | 4 | CYP5081 | A | 2 | 2 |
| CYP617 | A,B | 1,2 | 3 | CYP5125 | A | 2 | 2 |
| CYP5037 | B | 3 | 3 | CYP540 | B | 2 | 2 |
| CYP5110 | A | 3 | 3 | CYP630 | B | 2 | 2 |
| CYP530 | A | 3 | 3 | Others | - | - | 30 |
| Enzyme Family | KO Term | EC Number | Gene Number | Gene Name |
|---|---|---|---|---|
| 1,3-β-glucan synthase | K01180 | EC:3.2.1.6 | 1 | GI_10004256 |
| K00706 | EC:2.4.1.34 | 3 | GI_10014134, GI_10014600, GI_10010064 | |
| UTP–glucose-1-phosphate uridylyltransferase | K00963 | EC:2.7.7.9 | 3 | GI_10009949, GI_10009950, GI_10009951 |
| Hexokinase | K00844 | EC:2.7.1.1 | 2 | GI_10010509, GI_10009252 |
| Phosphoglucomutase | K01835 | EC:5.4.2.2 | 2 | GI_10003989, GI_10014463 |
| GTPase-activating-associated protein | K12492 | - | 1 | GI_10009154 |
| K19838 | - | 1 | GI_10009280 | |
| K12493 | - | 1 | GI_10004440 | |
| K14319 | - | 1 | GI_10004658 | |
| K19845 | - | 2 | GI_10004984, GI_10007354 | |
| K19839 | - | 3 | GI_10003462, GI_10010380, GI_10012746 | |
| K19844 | - | 2 | GI_10014590, GI_10000357 | |
| K18470 | - | 1 | GI_10014667 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Peng, J.; Sun, L.; Bonito, G.; Wang, J.; Cui, W.; Fu, Y.; Li, Y. Genome Sequencing Illustrates the Genetic Basis of the Pharmacological Properties of Gloeostereum incarnatum. Genes 2019, 10, 188. https://doi.org/10.3390/genes10030188
Wang X, Peng J, Sun L, Bonito G, Wang J, Cui W, Fu Y, Li Y. Genome Sequencing Illustrates the Genetic Basis of the Pharmacological Properties of Gloeostereum incarnatum. Genes. 2019; 10(3):188. https://doi.org/10.3390/genes10030188
Chicago/Turabian StyleWang, Xinxin, Jingyu Peng, Lei Sun, Gregory Bonito, Jie Wang, Weijie Cui, Yongping Fu, and Yu Li. 2019. "Genome Sequencing Illustrates the Genetic Basis of the Pharmacological Properties of Gloeostereum incarnatum" Genes 10, no. 3: 188. https://doi.org/10.3390/genes10030188
APA StyleWang, X., Peng, J., Sun, L., Bonito, G., Wang, J., Cui, W., Fu, Y., & Li, Y. (2019). Genome Sequencing Illustrates the Genetic Basis of the Pharmacological Properties of Gloeostereum incarnatum. Genes, 10(3), 188. https://doi.org/10.3390/genes10030188