Applying Genome-Resolved Metagenomics to Deconvolute the Halophilic Microbiome



Abstract
:1. Introduction
2. Shotgun Sequencing in Metagenomics
2.1. Halophilic Microbiome Research Powered by Shotgun Metagenomics
2.2. Limitations of Shotgun Metagenomics in Halophile Research
3. Experimental Design Considerations for Sequencing Halophilic Metagenomes
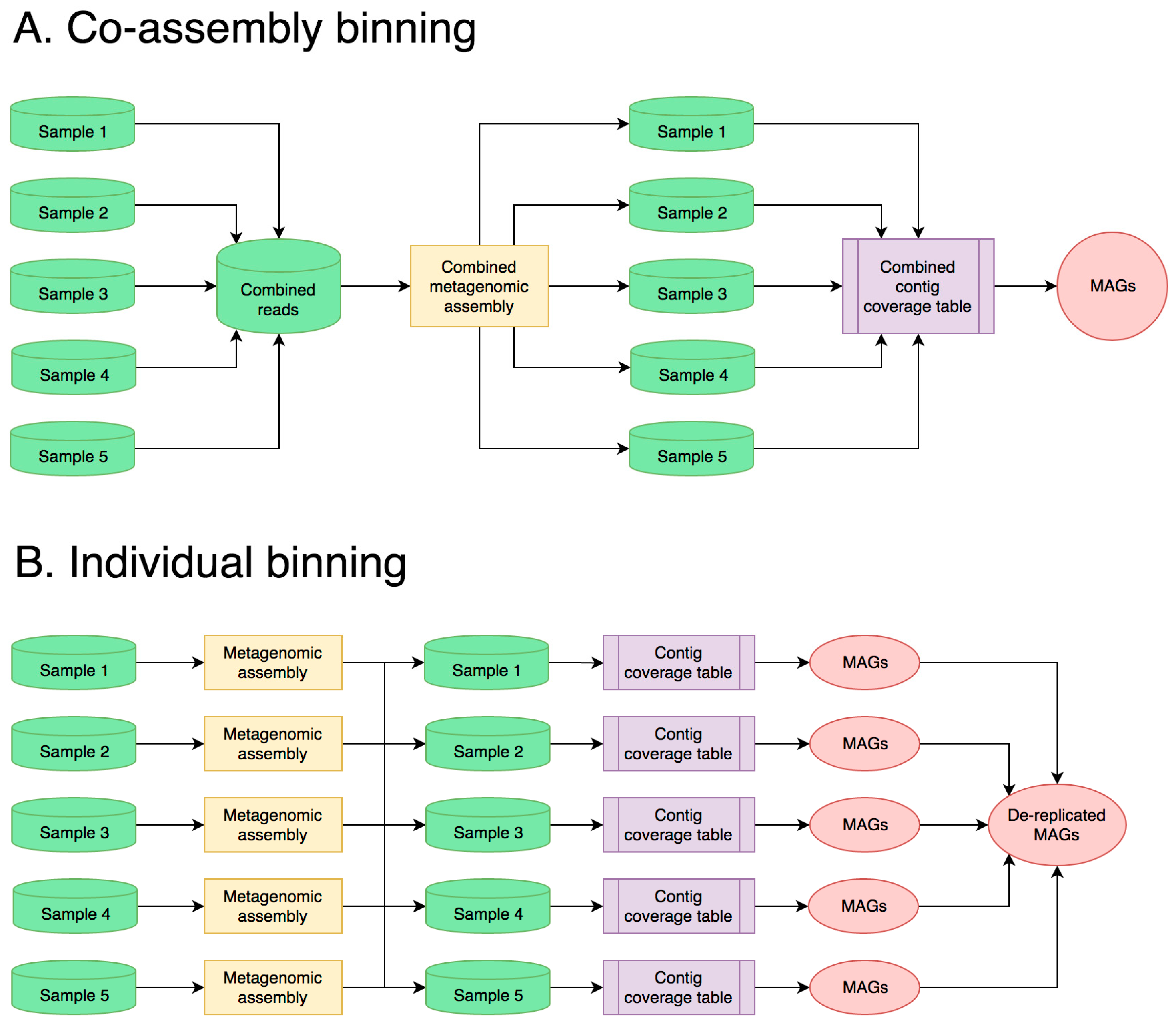
3.1. Best Bioinformatics Practices for Halophilic Metagenome Analysis
3.2. The Future of Halophilic Metagenomics
4. Conclusion
Acknowledgments
Conflicts of Interest
References
- Graham, E.B.; Knelman, J.E.; Schindlbacher, A.; Siciliano, S.; Breulmann, M.; Yannarell, A.; Beman, J.M.; Abell, G.; Philippot, L.; Prosser, J.; et al. Microbes as Engines of Ecosystem Function: When Does Community Structure Enhance Predictions of Ecosystem Processes? Front. Microbiol. 2016, 7, 111. [Google Scholar] [CrossRef] [PubMed]
- Kallmeyer, J.; Pockalny, R.; Adhikari, R.R.; Smith, D.C.; D’Hondt, S. Global distribution of microbial abundance and biomass in subseafloor sediment. Proc. Natl. Acad. Sci. USA 2012, 109, 16213–16216. [Google Scholar] [CrossRef] [PubMed]
- Whitman, W.B.; Coleman, D.C.; Wiebe, W.J. Prokaryotes: The unseen majority. Proc. Natl. Acad. Sci. USA 1998, 95, 6578–6583. [Google Scholar] [CrossRef] [PubMed]
- Vera-Gargallo, B.; Ventosa, A. Metagenomic Insights into the Phylogenetic and Metabolic Diversity of the Prokaryotic Community Dwelling in Hypersaline Soils from the Odiel Saltmarshes (SW Spain). Genes 2018, 9, 152. [Google Scholar] [CrossRef] [PubMed]
- Gibtan, A.; Park, K.; Woo, M.; Shin, J.-K.; Lee, D.-W.; Sohn, J.H.; Song, M.; Roh, S.W.; Lee, S.-J.; Lee, H.-S. Diversity of Extremely Halophilic Archaeal and Bacterial Communities from Commercial Salts. Front. Microbiol. 2017, 8, 631. [Google Scholar] [CrossRef] [PubMed]
- Henriet, O.; Fourmentin, J.; Delincé, B.; Mahillon, J. Exploring the diversity of extremely halophilic archaea in food-grade salts. Int. J. Food Microbiol. 2014, 191, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Seck, E.H.; Dufour, J.-C.; Raoult, D.; Lagier, J.-C. Halophilic & halotolerant prokaryotes in humans. Future Microbiol. 2018, 13, 799–812. [Google Scholar] [PubMed]
- Oren, A. Halophilic archaea on Earth and in space: Growth and survival under extreme conditions. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2014, 372, 20140194. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Galinski, E.A.; Grant, W.D.; Oren, A.; Ventosa, A. Halophiles 2010: Life in Saline Environments. Appl. Environ. Microbiol. 2010, 76, 6971–6981. [Google Scholar] [CrossRef] [PubMed]
- Rinke, C.; Schwientek, P.; Sczyrba, A.; Ivanova, N.N.; Anderson, I.J.; Cheng, J.-F.; Darling, A.; Malfatti, S.; Swan, B.K.; Gies, E.A.; et al. Insights into the phylogeny and coding potential of microbial dark matter. Nature 2013, 499, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Hedlund, B.P.; Dodsworth, J.A.; Murugapiran, S.K.; Rinke, C.; Woyke, T. Impact of single-cell genomics and metagenomics on the emerging view of extremophile “microbial dark matter.”. Extremophiles 2014, 18, 865–875. [Google Scholar] [CrossRef] [PubMed]
- Riesenfeld, C.S.; Schloss, P.D.; Handelsman, J. METAGENOMICS: Genomic Analysis of Microbial Communities. Annu. Rev. Genet. 2004, 38, 525–552. [Google Scholar] [CrossRef] [PubMed]
- Ranjan, R.; Rani, A.; Metwally, A.; McGee, H.S.; Perkins, D.L. Analysis of the microbiome: Advantages of whole genome shotgun versus 16S amplicon sequencing. Biochem. Biophys. Res. Commun. 2015, 469, 967–977. [Google Scholar] [CrossRef] [PubMed]
- Tessler, M.; Neumann, J.S.; Afshinnekoo, E.; Pineda, M.; Hersch, R.; Velho, L.F.M.; Segovia, B.T.; Lansac-Toha, F.A.; Lemke, M.; DeSalle, R.; et al. Large-scale differences in microbial biodiversity discovery between 16S amplicon and shotgun sequencing. Sci Rep. 2017, 7, 6589. [Google Scholar] [CrossRef] [PubMed]
- Quince, C.; Walker, A.W.; Simpson, J.T.; Loman, N.J.; Segata, N. Corrigendum: Shotgun metagenomics, from sampling to analysis. Nat. Biotechnol. 2017, 35, 1211. [Google Scholar] [CrossRef] [PubMed]
- Ghurye, J.S.; Cepeda-Espinoza, V.; Pop, M. Metagenomic Assembly: Overview, Challenges and Applications. Yale J. Biol Med. 2016, 89, 353–362. [Google Scholar] [PubMed]
- Olson, N.D.; Treangen, T.J.; Hill, C.M.; Cepeda-Espinoza, V.; Ghurye, J.; Koren, S.; Pop, M. Metagenomic assembly through the lens of validation: Recent advances in assessing and improving the quality of genomes assembled from metagenomes. Brief. Bioinform. 2017. [Google Scholar] [CrossRef] [PubMed]
- Uritskiy, G.V.; DiRuggiero, J.; Taylor, J. MetaWRAP—a flexible pipeline for genome-resolved metagenomic data analysis. Microbiome 2018, 6, 158. [Google Scholar] [CrossRef] [PubMed]
- Tully, B.J.; Graham, E.D.; Heidelberg, J.F. The reconstruction of 2,631 draft metagenome-assembled genomes from the global oceans. Sci. Data 2018, 5, 170203. [Google Scholar] [CrossRef] [PubMed]
- Sangwan, N.; Xia, F.; Gilbert, J.A. Recovering complete and draft population genomes from metagenome datasets. Microbiome 2016, 4, 197. [Google Scholar] [CrossRef] [PubMed]
- Sims, D.; Sudbery, I.; Ilott, N.E.; Heger, A.; Ponting, C.P. Sequencing depth and coverage: Key considerations in genomic analyses. Nat. Rev. Genet. 2014, 15, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Zaheer, R.; Noyes, N.; Polo, R.O.; Cook, S.R.; Marinier, E.; Van Domselaar, G.; Belk, K.E.; Morley, P.S.; McAllister, T.A. Impact of sequencing depth on the characterization of the microbiome and resistome. Sci. Rep. 2018, 8, 5890. [Google Scholar] [CrossRef] [PubMed]
- Sharifi, F.; Ye, Y. From Gene Annotation to Function Prediction for Metagenomics. Alcohol 2017, 1611, 27–34. [Google Scholar]
- Zhang, Y.; Wang, J.; Qi, J.; Zhao, F.; He, S.; Wei, S. Metagenomic sequencing reveals microbiota and its functional potential associated with periodontal disease. Sci. Rep. 2013, 3, 1843. [Google Scholar]
- Wang, W.-L.; Xu, S.-Y.; Ren, Z.-G.; Tao, L.; Jiang, J.-W.; Zheng, S.-S. Application of metagenomics in the human gut microbiome. WJG 2015, 21, 803–814. [Google Scholar] [CrossRef] [PubMed]
- Quince, C.; Walker, A.W.; Simpson, J.T.; Loman, N.J.; Segata, N. Shotgun metagenomics, from sampling to analysis. Nat. Biotechnol. 2017, 35, 833–844. [Google Scholar] [CrossRef] [PubMed]
- Poretsky, R.; Rodríguez-R, L.M.; Luo, C.; Tsementzi, D.; Konstantinidis, K.T. Strengths and Limitations of 16S rRNA Gene Amplicon Sequencing in Revealing Temporal Microbial Community Dynamics. PLoS ONE 2014, 9, e93827. [Google Scholar] [CrossRef] [PubMed]
- White, J.R.; Nagarajan, N.; Pop, M. Statistical Methods for Detecting Differentially Abundant Features in Clinical Metagenomic Samples. PLoS Comput. Biol. 2009, 5, e1000352. [Google Scholar] [CrossRef] [PubMed]
- Crits-Christoph, A.; Gelsinger, D.R.; Ma, B.; Wierzchos, J.; Ravel, J.; Davila, A.; Casero, M.C.; DiRuggiero, J. Functional interactions of archaea, bacteria, and viruses in a hypersaline endolithic community. Environm. Microbiol. 2016, 18, 2064–2077. [Google Scholar] [CrossRef] [PubMed]
- Naghoni, A.; Emtiazi, G.; Amoozegar, M.A.; Cretoiu, M.S.; Stal, L.J.; Etemadifar, Z.; Fazeli, S.A.S.; Bolhuis, H. Microbial diversity in the hypersaline Lake Meyghan, Iran. Sci. Rep. 2017, 7, 11522. [Google Scholar] [CrossRef] [PubMed]
- Plominsky, A.M.; Henríquez-Castillo, C.; Delherbe, N.; Podell, S.; Ramirez-Flandes, S.; Ugalde, J.A.; Santibañez, J.F.; Engh, G.V.D.; Hanselmann, K.; Ulloa, O.; et al. Distinctive Archaeal Composition of an Artisanal Crystallizer Pond and Functional Insights Into Salt-Saturated Hypersaline Environment Adaptation. Front. Microbiol. 2018, 9, 1800. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Enault, F.; Ravet, V.; Colombet, J.; Bettarel, Y.; Auguet, J.; Bouvier, T.; Lucas-Staat, S.; Vellet, A.; Prangishivili, D.; et al. Analysis of metagenomic data reveals common features of halophilic viral communities across continents. Environ. Microbiol. 2015, 18, 889–903. [Google Scholar] [CrossRef] [PubMed]
- Pedrós-Alió, C.; Calderón-Paz, J.I.; MacLean, M.H.; Medina, G.; Marrasé, C.; Gasol, J.M.; Guixa-Boixereu, N. The microbial food web along salinity gradients. FEMS Microbiol. Ecol. 2000, 32, 143–155. [Google Scholar] [CrossRef]
- Guixa-Boixareu, N.; Calderón-Paz, J.; Heldal, M.; Bratbak, G.; Pedrós-Alió, C. Viral lysis and bacterivory as prokaryotic loss factors along a salinity gradient. Aquat. Microb. Ecol. 1996, 11, 215–227. [Google Scholar] [CrossRef]
- Santos, F.; Meyerdierks, A.; Peña, A.; Amann, R.; Antón, J.; Rosselló-Mora, R.; Rosselló-Mora, R.; Rosselló-Móra, R. Metagenomic approach to the study of halophages: The environmental halophage 1. Environ. Microbiol. 2007, 9, 1711–1723. [Google Scholar] [CrossRef] [PubMed]
- Santos, F.; Yarza, P.; Parro, V.; Briones, C.; Antón, J. The metavirome of a hypersaline environment. Environ. Microbiol. 2010, 12, 2965–2976. [Google Scholar] [CrossRef] [PubMed]
- Antunes, A.; Alam, I.; Simões, M.F.; Daniels, C.; Ferreira, A.J.; Siam, R.; El-Dorry, H.; Bajic, V.B. First Insights into the Viral Communities of the Deep-sea Anoxic Brines of the Red Sea. Genomics Prot. Bioinform. 2015, 13, 304–309. [Google Scholar]
- Moller, A.G.; Liang, C. Determining virus-host interactions and glycerol metabolism profiles in geographically diverse solar salterns with metagenomics. PeerJ 2017, 5, e2844. [Google Scholar] [CrossRef] [PubMed]
- Finstad, K.M.; Probst, A.J.; Thomas, B.C.; Andersen, G.L.; Demergasso, C.; Echeverría, A.; Amundson, R.G.; Banfield, J.F. Microbial Community Structure and the Persistence of Cyanobacterial Populations in Salt Crusts of the Hyperarid Atacama Desert from Genome-Resolved Metagenomics. Front. Microbiol. 2017, 8, 1435. [Google Scholar] [CrossRef] [PubMed]
- Kimbrel, J.A.; Ballor, N.; Wu, Y.-W.; David, M.M.; Hazen, T.C.; Simmons, B.A.; Singer, S.W.; Jansson, J.K. Microbial Community Structure and Functional Potential Along a Hypersaline Gradient. Front. Microbiol. 2018, 9, 1492. [Google Scholar] [CrossRef] [PubMed]
- Uritskiy, G.; Getsin, S.; Munn, A.; Gomez-Silva, B.; Davila, A.; Glass, B.; Taylor, J.; DiRuggiero, J. Response of extremophile microbiome to a rare rainfall reveals a two-step adaptation mechanism. bioRxiv 2018, bioRxiv, 442525. [Google Scholar]
- Becker, E.A.; Seitzer, P.M.; Tritt, A.; Larsen, D.; Krusor, M.; Yao, A.I.; Wu, D.; Madern, D.; Eisen, J.A.; Darling, A.E.; et al. Phylogenetically Driven Sequencing of Extremely Halophilic Archaea Reveals Strategies for Static and Dynamic Osmo-response. PLoS Genet. 2014, 10, e1004784. [Google Scholar] [CrossRef] [PubMed]
- DeMaere, M.Z.; Williams, T.J.; Allen, M.A.; Brown, M.V.; Gibson, J.A.E.; Rich, J.; Lauro, F.M.; Dyall-Smith, M.; Davenport, K.W.; Woyke, T.; et al. High level of intergenera gene exchange shapes the evolution of haloarchaea in an isolated Antarctic lake. Proc. Natl. Acad. Sci. USA 2013, 110, 16939–16944. [Google Scholar] [CrossRef] [PubMed]
- Tschitschko, B.; Erdmann, S.; Roux, S.; Panwar, P.; Brazendale, S.; Cavicchioli, R.; DeMaere, M.Z.; A Allen, M.; Williams, T.J.; Hancock, A.M.; et al. Genomic variation and biogeography of Antarctic haloarchaea. Microbiome 2018, 6, 113. [Google Scholar] [CrossRef] [PubMed]
- Mobberley, J.M.; Lindemann, S.R.; Bernstein, H.C.; Moran, J.J.; Renslow, R.S.; Babauta, J.; Hu, D.; Beyenal, H.; Nelson, W.C. Organismal and spatial partitioning of energy and macronutrient transformations within a hypersaline mat. FEMS Microbiol. Ecol. 2017, 93. [Google Scholar] [CrossRef] [PubMed]
- Loukas, A.; Kappas, I.; Abatzopoulos, T.J. HaloDom: A new database of halophiles across all life domains. J. Biol. Res. Thessalon. 2018, 25, 2. [Google Scholar] [CrossRef] [PubMed]
- Solden, L.; Lloyd, K.; Wrighton, K. The bright side of microbial dark matter: Lessons learned from the uncultivated majority. Curr. Opin. Microbiol. 2016, 31, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Ventosa, A.; De La Haba, R.R.; Sánchez-Porro, C.; Papke, R.T. Microbial diversity of hypersaline environments: A metagenomic approach. Curr. Opin. Microbiol. 2015, 25, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Narasingarao, P.; Podell, S.; A Ugalde, J.; Brochier-Armanet, C.; Emerson, J.B.; Brocks, J.J.; Heidelberg, K.B.; Banfield, J.F.; E Allen, E. De novo metagenomic assembly reveals abundant novel major lineage of Archaea in hypersaline microbial communities. ISME J. 2011, 6, 81–93. [Google Scholar] [CrossRef] [PubMed]
- Pašić, L.; Fernández, A.B.; Martin-Cuadrado, A.-B.; Papke, R.T.; Rodriguez-Brito, B.; Ghai, R.; Mizuno, C.M.; McMahon, K.D.; Stepanauskas, R.; Rohwer, F.; et al. New Abundant Microbial Groups in Aquatic Hypersaline Environments. Sci. Rep. 2011, 1, 135. [Google Scholar]
- Andrade, K.; Logemann, J.; Heidelberg, K.B.; Emerson, J.B.; Comolli, L.R.; A Hug, L.; Probst, A.J.; Keillar, A.; Thomas, B.C.; Miller, C.S.; et al. Metagenomic and lipid analyses reveal a diel cycle in a hypersaline microbial ecosystem. ISME J. 2015, 9, 2697–2711. [Google Scholar] [CrossRef] [PubMed]
- Podell, S.; Ugalde, J.A.; Narasingarao, P.; Banfield, J.F.; Heidelberg, K.B.; Allen, E.E. Assembly-Driven Community Genomics of a Hypersaline Microbial Ecosystem. PLoS ONE 2013, 8, e61692. [Google Scholar] [CrossRef] [PubMed]
- Vavourakis, C.D.; Andrei, A.-S.; Mehrshad, M.; Ghai, R.; Sorokin, D.Y.; Muyzer, G. A metagenomics roadmap to the uncultured genome diversity in hypersaline soda lake sediments. Microbiome 2018, 6, 168. [Google Scholar] [CrossRef] [PubMed]
- Emerson, J.B.; Andrade, K.; Thomas, B.C.; Norman, A.; Allen, E.E.; Heidelberg, K.B.; Banfield, J.F. Virus-host and CRISPR dynamics in Archaea-dominated hypersaline Lake Tyrrell, Victoria, Australia. Archaea 2013, 370871. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Barbero, M.D.; Martínez, J.M.; Almansa, C.; Rodríguez, N.; Villamor, J.; Gomariz, M.; Escudero, C.; Rubin, S.; Antón, J.; Martínez-García, M.; et al. Prokaryotic and viral community structure in the singular chaotropic salt lake salar de uyuni. Environ. Microbiol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Barbero, M.D.; Martin-Cuadrado, A.-B.; Viver, T.; Santos, F.; Martinez-Garcia, M.; Antón, J. Recovering microbial genomes from metagenomes in hypersaline environments: The Good, the Bad and the Ugly. Syst. Appl. Microbiol. 2019, 42, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Di Meglio, L.; Santos, F.; Gomariz, M.; Almansa, C.; López, C.; Antón, J.; Nercessian, D. Seasonal dynamics of extremely halophilic microbial communities in three Argentinian salterns. FEMS Microbiol. Ecol. 2016, 92, fiw184. [Google Scholar] [CrossRef] [PubMed]
- Berlanga, M.; Palau, M.; Guerrero, R. Functional Stability and Community Dynamics during Spring and Autumn Seasons Over 3 Years in Camargue Microbial Mats. Front. Microbiol. 2017, 8, 2619. [Google Scholar] [CrossRef] [PubMed]
- Ruvindy, R.; White, R.A., 3rd; Neilan, B.A..; Burns, B.P. Unravelling core microbial metabolisms in the hypersaline microbial mats of Shark Bay using high-throughput metagenomics. ISME J. 2016, 10, 183–196. [Google Scholar] [CrossRef] [PubMed]
- Wong, H.L.; White, R.A.; Visscher, P.T.; Charlesworth, J.C.; Vázquez-Campos, X.; Burns, B.P. Disentangling the drivers of functional complexity at the metagenomic level in Shark Bay microbial mat microbiomes. ISME J. 2018, 12, 2619–2639. [Google Scholar] [CrossRef] [PubMed]
- White, R.A.; Wong, H.L.; Ruvindy, R.; Neilan, B.A.; Burns, B.P.; Iii, R.A.W. Viral Communities of Shark Bay Modern Stromatolites. Front. Microbiol. 2018, 9, 1223. [Google Scholar] [CrossRef] [PubMed]
- Speth, D.R.; Lagkouvardos, I.; Wang, Y.; Qian, P.-Y.; Dutilh, B.E.; Jetten, M.S.M. Draft Genome of Scalindua rubra, Obtained from the Interface Above the Discovery Deep Brine in the Red Sea, Sheds Light on Potential Salt Adaptation Strategies in Anammox Bacteria. Microb. Ecol. 2017, 74, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; Hikmawan, T.; Antunes, A.; Ngugi, D.; Stingl, U. Diversity of methanogens and sulfate-reducing bacteria in the interfaces of five deep-sea anoxic brines of the Red Sea. Res. Microbiol. 2015, 166, 688–699. [Google Scholar] [CrossRef] [PubMed]
- Pachiadaki, M.G.; Yakimov, M.M.; Lacono, V.; Leadbetter, E.; Edgcomb, V. Unveiling microbial activities along the halocline of Thetis, a deep-sea hypersaline anoxic basin. ISME J. 2014, 8, 2478–2489. [Google Scholar] [CrossRef] [PubMed]
- Crits-Christoph, A.; Robinson, C.K.; Ma, B.; Ravel, J.; Wierzchos, J.; Ascaso, C.; Artieda, O.; Souza-Egipsy, V.; Casero, M.C.; DiRuggiero, J. Phylogenetic and Functional Substrate Specificity for Endolithic Microbial Communities in Hyper-Arid Environments. Front. Microbiol. 2016, 7, 301. [Google Scholar] [CrossRef] [PubMed]
- Narayan, A.; Patel, V.; Singh, P.; Patel, A.; Jain, K.; Karthikeyan, K.; Shah, A.; Madamwar, D. Response of microbial community structure to seasonal fluctuation on soils of Rann of Kachchh, Gujarat, India: Representing microbial dynamics and functional potential. Ecol. Genet. Genomics 2018, 6, 22–32. [Google Scholar] [CrossRef]
- Pandit, A.S.; Joshi, M.N.; Bhargava, P.; Shaikh, I.; Ayachit, G.N.; Raj, S.R.; Saxena, A.K.; Bagatharia, S.B. A snapshot of microbial communities from the Kutch: One of the largest salt deserts in the World. Extremophiles 2015, 19, 973–987. [Google Scholar] [CrossRef] [PubMed]
- Cuadros-Orellana, S.; Martin-Cuadrado, A.-B.; Legault, B.; D’Auria, G.; Zhaxybayeva, O.; Papke, R.T.; Rodriguez-Valera, F. Genomic plasticity in prokaryotes: The case of the square haloarchaeon. ISME J. 2007, 1, 235–245. [Google Scholar] [CrossRef] [PubMed]
- Papke, R.T.; Koenig, J.E.; Rodriguez-Valera, F.; Doolittle, W.F. Frequent Recombination in a Saltern Population of Halorubrum. Science 2004, 306, 1928–1929. [Google Scholar] [PubMed]
- Pašić, L.; Rodriguez-Mueller, B.; Martin-Cuadrado, A.-B.; Rodriguez-Valera, F.; Mira, A.; Rohwer, F. Metagenomic islands of hyperhalophiles: The case of Salinibacter ruber. BMC Genomics 2009, 10, 570. [Google Scholar] [CrossRef] [PubMed]
- Martin-Cuadrado, A.-B.; Pašić, L.; Rodriguez-Valera, F. Diversity of the cell-wall associated genomic island of the archaeon Haloquadratum walsbyi. BMC Genomics 2015, 16, 13950. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-C.; Liu, T.; Yu, C.-H.; Chiang, T.-Y.; Hwang, C.-C. Effects of GC bias in next-generation-sequencing data on de novo genome assembly. PLoS ONE 2013, 8, e62856. [Google Scholar] [CrossRef] [PubMed]
- Haft, D.H.; DiCuccio, M.; Badretdin, A.; Brover, V.; Chetvernin, V.; O’Neill, K.; Li, W.; Chitsaz, F.; Derbyshire, M.K.; Gonzales, N.R.; et al. RefSeq: An update on prokaryotic genome annotation and curation. Nucleic Acids Res. 2017, 46, D851–D860. [Google Scholar] [CrossRef] [PubMed]
- Nurk, S.; Meleshko, D.; Korobeynikov, A.; Pevzner, P.A. metaSPAdes: A new versatile metagenomic assembler. Genome Res. 2017, 27, 824–834. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.D.; Froula, J.; Egan, R.; Wang, Z.; Rahmann, S. MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. PeerJ 2015, 3, 1165. [Google Scholar] [CrossRef] [PubMed]
- Haro-Moreno, J.M.; López-Pérez, M.; De La Torre, J.R.; Picazo, A.; Camacho, A.; Rodriguez-Valera, F. Fine metagenomic profile of the Mediterranean stratified and mixed water columns revealed by assembly and recruitment. Microbiome 2018, 6, 128. [Google Scholar] [CrossRef] [PubMed]
- Olm, M.R.; Brown, C.T.; Brooks, B.; Banfield, J.F. dRep: A tool for fast and accurate genomic comparisons that enables improved genome recovery from metagenomes through de-replication. ISME J. 2017, 11, 2864–2868. [Google Scholar] [CrossRef] [PubMed]
- Goodrich, J.K.; Di Rienzi, S.C.; Poole, A.C.; Koren, O.; Walters, W.A.; Caporaso, J.G.; Knight, R.; Ley, R.E. Conducting a microbiome study. Cell 2014, 158, 250–262. [Google Scholar] [CrossRef] [PubMed]
- Vavourakis, C.D.; Ghai, R.; Rodríguez-Valera, F.; Sorokin, D.Y.; Tringe, S.G.; Hugenholtz, P.; Muyzer, G. Metagenomic Insights into the Uncultured Diversity and Physiology of Microbes in Four Hypersaline Soda Lake Brines. Front. Microbiol. 2016, 7, 533. [Google Scholar] [CrossRef] [PubMed]
- Stewart, R.D.; Auffret, M.D.; Warr, A.; Wiser, A.H.; Press, M.O.; Langford, K.W.; Liachko, I.; Snelling, T.J.; Dewhurst, R.J.; Walker, A.W.; et al. Assembly of 913 microbial genomes from metagenomic sequencing of the cow rumen. Nat. Commun 2018, 9, 870. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, S.M.; Duvallet, C.; Alm, E.J. Correcting for batch effects in case-control microbiome studies. PLoS Comput. Biol. 2018, 14, e1006102. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.; Bag, S.K.; Das, S.; Harvill, E.T.; Dutta, C. Molecular signature of hypersaline adaptation: Insights from genome and proteome composition of halophilic prokaryotes. Genome Biol. 2008, 9. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.B.; Highlander, S.K.; Anderson, E.L.; Li, W.; Dayrit, M.; Klitgord, N.; Fabani, M.M.; Seguritan, V.; Green, J.; Pride, D.T.; et al. Library preparation methodology can influence genomic and functional predictions in human microbiome research. Proc. Natl. Acad. Sci. USA 2015, 112, 14024–14029. [Google Scholar] [CrossRef] [PubMed]
- Tamames, J.; Puente-Sanchez, F. SqueezeM, a highly portable, fully automatic metagenomic analysis pipeline. Bioinformatics 2018, bioRxiv, 347559. [Google Scholar]
- Brown, J.; Pirrung, M.; McCue, L.A. FQC Dashboard: Integrates FastQC results into a web-based, interactive, and extensible FASTQ quality control tool. Bioinformatics 2017, 33, 3137–3139. [Google Scholar] [CrossRef] [PubMed]
- Mikheenko, A.; Saveliev, V.; Gurevich, A. MetaQUAST: Evaluation of metagenome assemblies. Bioinformatics 2015, 32, 1088–1090. [Google Scholar] [CrossRef] [PubMed]
- Sczyrba, A.; Hofmann, P.; Belmann, P.; Koslicki, D.; Janssen, S.; Dröge, J.; Gregor, I.; Majda, S.; Fiedler, J.; Dahms, E.; et al. Critical Assessment of Metagenome Interpretation-a benchmark of metagenomics software. Nat. Methods 2017, 14, 1063–1071. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Luo, R.; Liu, C.-M.; Leung, C.-M.; Ting, H.-F.; Sadakane, K.; Yamashita, H.; Lam, T.-W. MEGAHIT v1.0: A fast and scalable metagenome assembler driven by advanced methodologies and community practices. Methods 2016, 102, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Vollmers, J.; Wiegand, S.; Kaster, A.-K. Comparing and Evaluating Metagenome Assembly Tools from a Microbiologist’s Perspective—Not Only Size Matters! PLoS ONE 2017, 12, e0169662. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, D.L. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2001, 29, 11–16. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2015, 44, D733–D745. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Ye, W.; Zhang, Y.; Xu, Y. High speed BLASTN: An accelerated MegaBLAST search tool. Nucleic Acids Res. 2015, 43, 7762–7768. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.S.; Baker, B.J.; Thomas, B.C.; Singer, S.W.; Banfield, J.F. EMIRGE: Reconstruction of full-length ribosomal genes from microbial community short read sequencing data. Genome Biol. 2011, 12, R44. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glockner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.A.; Markowitz, V.M.; Chu, K.; Palaniappan, K.; Szeto, E.; Pillay, M.; Ratner, A.; Huang, J.; Andersen, E.; Huntemann, M.; et al. IMG/M: Integrated genome and metagenome comparative data analysis system. Nucleic Acids. Res. 2017, 45, D507–D516. [Google Scholar] [CrossRef] [PubMed]
- Abubucker, S.; Segata, N.; Goll, J.; Schubert, A.M.; Izard, J.; Cantarel, B.L.; Rodriguez-Mueller, B.; Zucker, J.; Thiagarajan, M.; Henrissat, B.; et al. Metabolic Reconstruction for Metagenomic Data and Its Application to the Human Microbiome. PLoS Comput. Biol. 2012, 8, e1002358. [Google Scholar] [CrossRef] [PubMed]
- Alneberg, J.; Bjarnason, B.S.; De Bruijn, I.; Schirmer, M.; Quick, J.; Ijaz, U.Z.; Lahti, L.; Loman, N.J.; Andersson, A.F.; Quince, C. Binning metagenomic contigs by coverage and composition. Nat. Methods 2014, 11, 1144–1146. [Google Scholar] [CrossRef] [PubMed]
- Sieber, C.M.K.; Probst, A.J.; Sharrar, A.; Thomas, B.C.; Hess, M.; Tringe, S.G.; Banfield, J.F. Recovery of genomes from metagenomes via a dereplication, aggregation and scoring strategy. Nat. Microbiol. 2018, 3, 836–843. [Google Scholar] [CrossRef] [PubMed]
- Wommack, K.E.; Bhavsar, J.; Ravel, J. Metagenomics: Read length matters. Appl. Environ. Microbiol. 2008, 74, 1453–1463. [Google Scholar] [CrossRef] [PubMed]
- Rhoads, A.; Au, K.F. PacBio Sequencing and Its Applications. Genomics Prot. Bioinform. 2015, 13, 278–289. [Google Scholar]
- Frank, J.A.; Pan, Y.; Tooming-Klunderud, A.; Eijsink, V.G.H.; McHardy, A.C.; Nederbragt, A.J.; Pope, P.B.; Nederbragt, A.; Pope, P. Improved metagenome assemblies and taxonomic binning using long-read circular consensus sequence data. Sci. Rep. 2016, 6, 25373. [Google Scholar] [CrossRef] [PubMed]
- Brown, B.L.; Watson, M.; Minot, S.S.; Rivera, M.C.; Franklin, R.B. MinION™ nanopore sequencing of environmental metagenomes: A synthetic approach. GigaScience 2017, 6, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Rang, F.J.; Kloosterman, W.P.; De Ridder, J. From squiggle to basepair: Computational approaches for improving nanopore sequencing read accuracy. Genome Biol. 2018, 19, 90. [Google Scholar] [CrossRef] [PubMed]
- Driscoll, C.B.; Otten, T.G.; Brown, N.M.; Dreher, T.W. Towards long-read metagenomics: Complete assembly of three novel genomes from bacteria dependent on a diazotrophic cyanobacterium in a freshwater lake co-culture. Stand. Genomic Sci. 2017, 12, 9. [Google Scholar] [CrossRef] [PubMed]
- Moss, E.; Bishara, A.; Tkachenko, E.; Kang, J.B.; Andermann, T.M.; Wood, C.; Handy, C.; Ji, H.; Batzoglou, S.; Bhatt, A.S. De novo assembly of microbial genomes from human gut metagenomes using barcoded short read sequences. bioRxiv 2017. [Google Scholar] [CrossRef]
- Jain, M.; Koren, S.; Miga, K.H.; Quick, J.; Rand, A.C.; A Sasani, T.; Tyson, J.R.; Beggs, A.D.; Dilthey, A.T.; Fiddes, I.T.; et al. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat. Biotechnol 2018, 36, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Press, M.O.; Wiser, A.H.; Kronenberg, Z.N.; Langford, K.W.; Shakya, M.; Lo, C.-C.; Mueller, K.A.; Sullivan, S.T.; Chain, P.S.G.; Liachko, I. Hi-C deconvolution of a human gut microbiome yields high-quality draft genomes and reveals plasmid-genome interactions. Genomics 2017, bioRxiv, 198713. [Google Scholar]
- Burton, J.N.; Liachko, I.; Dunham, M.J.; Shendure, J. Species-Level Deconvolution of Metagenome Assemblies with Hi-C–Based Contact Probability Maps. G3 2014, 4, 1339–1346. [Google Scholar] [CrossRef] [PubMed]
- Lavelle, A.; Sokol, H. Gut microbiota: Beyond metagenomics, metatranscriptomics illuminates microbiome functionality in IBD. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 193–194. [Google Scholar] [CrossRef] [PubMed]
- Ren, M.; Zhang, Z.; Wang, X.; Zhou, Z.; Chen, D.; Zeng, H.; Zhao, S.; Chen, L.; Hu, Y.; Zhang, C.; et al. Diversity and Contributions to Nitrogen Cycling and Carbon Fixation of Soil Salinity Shaped Microbial Communities in Tarim Basin. Front. Microbiol. 2018, 9, 431. [Google Scholar] [CrossRef] [PubMed]
- Garoutte, A.; Cardenas, E.; Tiedje, J.; Howe, A. Methodologies for probing the metatranscriptome of grassland soil. J. Microbiol. Methods 2016, 131, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Xiong, X.; Danska, J.; Parkinson, J. Metatranscriptomic analysis of diverse microbial communities reveals core metabolic pathways and microbiome-specific functionality. Microbiome 2016, 4, 13780. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Environment | Longitudinal Dynamics | MAG Discovery | Functional Potential | Virus Analysis |
|---|---|---|---|---|
| Hypersaline lakes | Andrade [51], Tschitschko [44], Podell [52] | Narasingarao [49] | Vavourakis [53], Naghoni [30] | Emerson [54], Tschitschko [44], Ramos-Barbero [55] |
| Salterns | Plominsky [2] | Ramos-Barbero [56], Ghai [50] | Plominsky [31], Ghai [50] | Moller [38], Di Meglio [57] |
| Hypersaline microbial mats | Mobberley [45], Berlanga [58] | Mobberley [45] | Mobberley [45], Ruvindy [59], Wong [60] | White [61] |
| Haloclines | N/A | Speth [62] | Guan [63], Pachiadaki [64] | Antunes [37] |
| Halite endoliths | Uritskiy [41], Finstad [39] | Finstad [39], Uritskiy [41], | Crits-Christoph [65], Uritskiy [41] | Crits-Christoph [65] |
| Hypersaline soils | Narayan [66] | Vera-Gargallo [4] | Vera-Gargallo [4], Pandit [67] | NA |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Uritskiy, G.; DiRuggiero, J. Applying Genome-Resolved Metagenomics to Deconvolute the Halophilic Microbiome. Genes 2019, 10, 220. https://doi.org/10.3390/genes10030220
Uritskiy G, DiRuggiero J. Applying Genome-Resolved Metagenomics to Deconvolute the Halophilic Microbiome. Genes. 2019; 10(3):220. https://doi.org/10.3390/genes10030220
Chicago/Turabian StyleUritskiy, Gherman, and Jocelyne DiRuggiero. 2019. "Applying Genome-Resolved Metagenomics to Deconvolute the Halophilic Microbiome" Genes 10, no. 3: 220. https://doi.org/10.3390/genes10030220
APA StyleUritskiy, G., & DiRuggiero, J. (2019). Applying Genome-Resolved Metagenomics to Deconvolute the Halophilic Microbiome. Genes, 10(3), 220. https://doi.org/10.3390/genes10030220