The Primary Antisense Transcriptome of Halobacterium salinarum NRC-1

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Antisense Transcription Start Sites Annotation
2.2. Antisense RNA Loci Inference
2.3. Promoter and 3′ End Sequence Analysis
2.4. Gene Functions
2.5. Type II Toxin-Antitoxin Systems Annotation and Antisense Transcription Start Sites Identification in Other Archaea
2.6. Differential Expression Analysis
2.7. Antisense Transcription Start Sites Comparison between Halobacterium salinarum and Haloferax volcanii
2.8. Ribo-Seq Data Analysis
3. Results
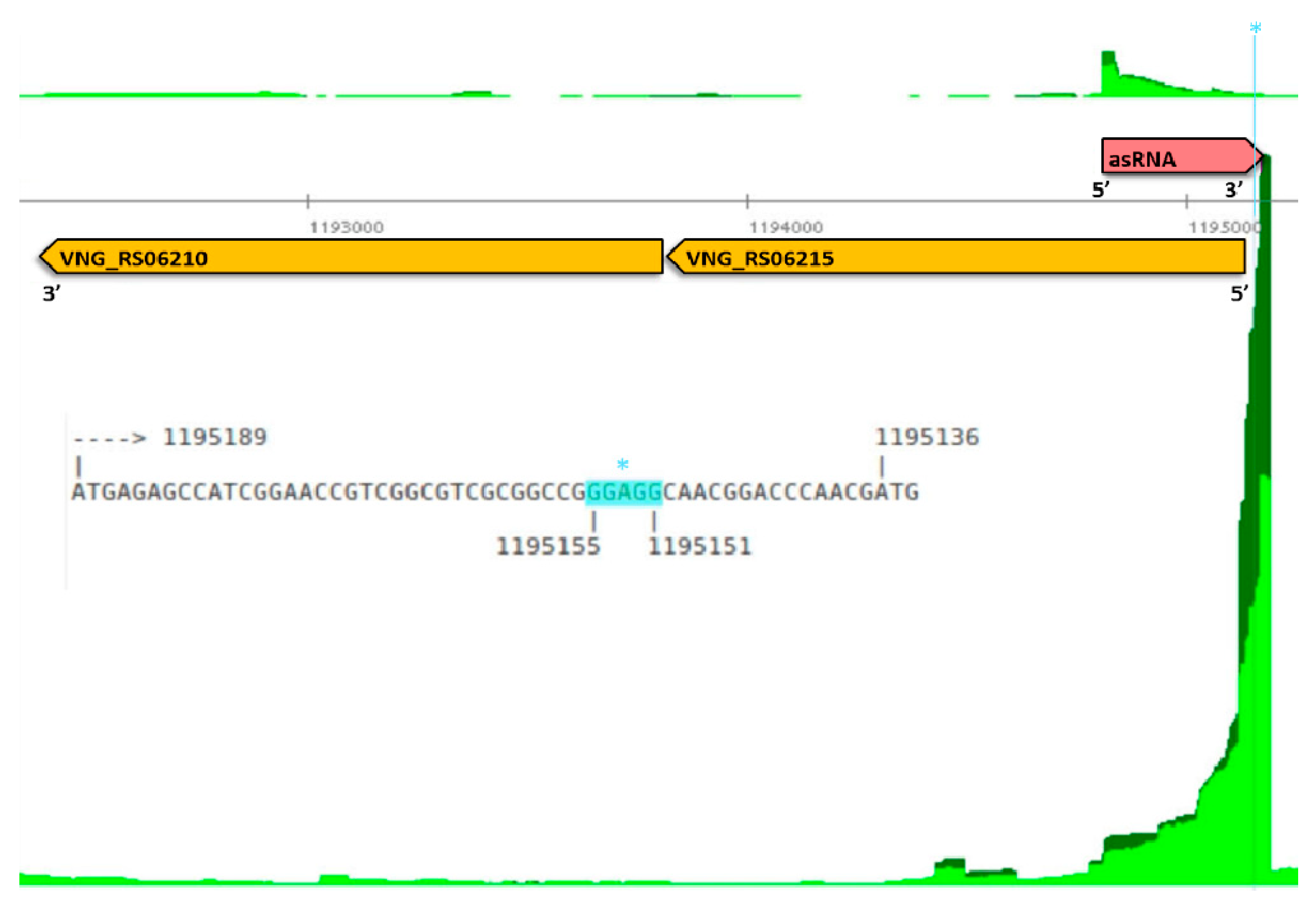
3.1. Mapping Primary Antisense RNAs
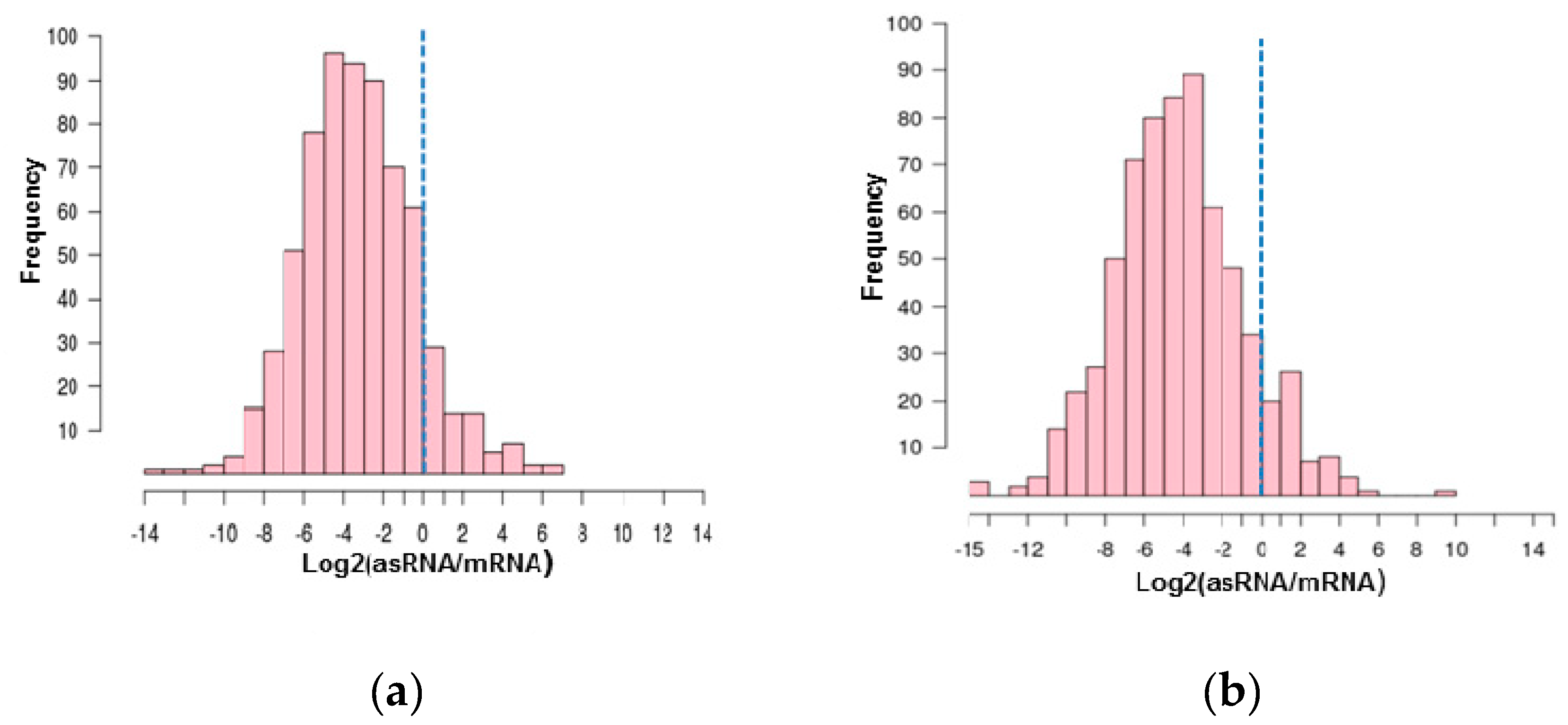
3.2. Antisense RNAs Expression Levels
3.3. Function of the Genes on the Opposite Strand of Antisense RNAs
3.4. Ribosome-Associated Antisense RNAs
3.5. Conservation of Antisense Transcription Start Sites
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sayed, N.; Jousselin, A.; Felden, B. A cis-antisense RNA acts in trans in Staphylococcus aureus to control translation of a human cytolytic peptide. Nat. Struct. Mol. Biol. 2012, 19, 105–113. [Google Scholar] [CrossRef]
- Pelechano, V.; Steinmetz, L.M. Gene regulation by antisense transcription. Nat. Rev. Genet. 2013, 14, 880–893. [Google Scholar] [CrossRef]
- Lasa, I.; Toledo-Arana, A.; Dobin, A.; Villanueva, M.; de Los Mozos, I.R.; Vergara-Irigaray, M.; Segura, V.; Fagegaltier, D.; Penadés, J.R.; Valle, J.; et al. Genome-wide antisense transcription drives mRNA processing in bacteria. Proc. Natl. Acad. Sci. USA 2011, 108, 20172–20177. [Google Scholar] [CrossRef]
- Kawano, M.; Aravind, L.; Storz, G. An antisense RNA controls synthesis of an SOS-induced toxin evolved from an antitoxin. Mol. Microbiol. 2007, 64, 738–754. [Google Scholar] [CrossRef]
- Carrieri, C.; Cimatti, L.; Biagioli, M.; Beugnet, A.; Zucchelli, S.; Fedele, S.; Pesce, E.; Ferrer, I.; Collavin, L.; Santoro, C.; et al. Long non-coding antisense RNA controls Uchl1 translation through an embedded SINEB2 repeat. Nature 2012, 491, 454–459. [Google Scholar] [CrossRef]
- Bøvre, K.; Szybalski, W. Patterns of convergent and overlapping transcription within the b2 region of coliphage λ. Virology 1969, 38, 614–626. [Google Scholar] [CrossRef]
- Inouye, M. Antisense RNA: Its functions and applications in gene regulation—A review. Gene 1988, 72, 25–34. [Google Scholar] [CrossRef]
- Vanhée-Brossollet, C.; Vaquero, C. Do natural antisense transcripts make sense in eukaryotes? Gene 1998, 211, 1–9. [Google Scholar] [CrossRef]
- Lasa, I.; Toledo-Arana, A.; Gingeras, T. An effort to make sense of antisense transcription in bacteria. RNA Biol. 2012, 9, 1039–1044. [Google Scholar] [CrossRef]
- Levin, J.Z.; Yassour, M.; Adiconis, X.; Nusbaum, C.; Thompson, D.A.; Friedman, N.; Gnirke, A.; Regev, A. Comprehensive comparative analysis of strand-specific RNA sequencing methods. Nat. Methods 2010, 7, 709–715. [Google Scholar] [CrossRef]
- Sharma, C.M.; Vogel, J. Differential RNA-seq: The approach behind and the biological insight gained. Curr. Opin. Microbiol. 2014, 19, 97–105. [Google Scholar] [CrossRef]
- Sun, Y.; Li, D.; Zhang, R.; Peng, S.; Zhang, G.; Yang, T.; Qian, A. Strategies to identify natural antisense transcripts. Biochimie 2017, 132, 131–151. [Google Scholar] [CrossRef]
- Beiter, T.; Reich, E.; Williams, R.W.; Simon, P. Antisense transcription: A critical look in both directions. Cell. Mol. Life Sci. 2009, 66, 94–112. [Google Scholar] [CrossRef]
- Georg, J.; Hess, W.R. Cis-antisense RNA, another level of gene regulation in bacteria. Microbiol. Mol. Biol. Rev. 2011, 75, 286–300. [Google Scholar] [CrossRef]
- Wade, J.T.; Grainger, D.C. Pervasive transcription: Illuminating the dark matter of bacterial transcriptomes. Nat. Rev. Microbiol. 2014, 12, 647–653. [Google Scholar] [CrossRef]
- Lloréns-Rico, V.; Cano, J.; Kamminga, T.; Gil, R.; Latorre, A.; Chen, W.H.; Bork, P.; Glass, J.I.; Serrano, L.; Lluch-Senar, M. Bacterial antisense RNAs are mainly the product of transcriptional noise. Sci. Adv. 2016, 2, e1501363. [Google Scholar] [CrossRef]
- Wagner, E.G.H.; Romby, P. Chapter three-small RNAs in bacteria and archaea: Who they are, what they do, and how they do it. Adv. Genet. 2015, 90, 133–208. [Google Scholar]
- Eckweiler, D.; Häussler, S. Antisense transcription in Pseudomonas aeruginosa. Microbiology 2018, 164, 889–895. [Google Scholar] [CrossRef]
- Krüger, K.; Pfeifer, F. Transcript analysis of the c-vac region and differential synthesis of the two regulatory gas vesicle proteins GvpD and GvpE in Halobacterium salinarium PHH4. J. Bacteriol. 1996, 178, 4012–4019. [Google Scholar] [CrossRef]
- Gelsinger, D.R.; DiRuggiero, J. The non-coding regulatory RNA revolution in archaea. Genes 2018, 9, 141. [Google Scholar] [CrossRef]
- Babski, J.; Haas, K.A.; Näther-Schindler, D.; Pfeiffer, F.; Förstner, K.U.; Hammelmann, M.; Hilker, R.; Becker, A.; Sharma, C.M.; Marchfelder, A.; et al. Genome-wide identification of transcriptional start sites in the haloarchaeon Haloferax volcanii based on differential RNA-Seq (dRNA-Seq). BMC Genom. 2016, 17, 629. [Google Scholar] [CrossRef]
- Li, J.; Qi, L.; Guo, Y.; Yue, L.; Li, Y.; Ge, W.; Wu, J.; Shi, W.; Dong, X. Global mapping transcriptional start sites revealed both transcriptional and post-transcriptional regulation of cold adaptation in the methanogenic archaeon Methanolobus psychrophilus. Sci. Rep. 2015, 5, 9209. [Google Scholar] [CrossRef] [PubMed]
- Jäger, D.; Förstner, K.U.; Sharma, C.M.; Santangelo, T.J.; Reeve, J.N. Primary transcriptome map of the hyperthermophilic archaeon Thermococcus kodakarensis. BMC Genom. 2014, 15, 684. [Google Scholar] [CrossRef] [PubMed]
- Jäger, D.; Sharma, C.M.; Thomsen, J.; Ehlers, C.; Vogel, J.; Schmitz, R.A. Deep sequencing analysis of the Methanosarcina mazei Go1 transcriptome in response to nitrogen availability. Proc. Natl. Acad. Sci. USA 2009, 106, 21878–21882. [Google Scholar] [CrossRef]
- Cho, S.; Kim, M.-S.; Jeong, Y.; Lee, B.-R.; Lee, J.-H.; Kang, S.G.; Cho, B.-K. Genome-wide primary transcriptome analysis of H2-producing archaeon Thermococcus onnurineus NA1. Sci. Rep. 2017, 7, 43044. [Google Scholar] [CrossRef]
- Smollett, K.; Blombach, F.; Reichelt, R.; Thomm, M.; Werner, F. A global analysis of transcription reveals two modes of Spt4/5 recruitment to archaeal RNA polymerase. Nat. Microbiol. 2017, 2, 17021. [Google Scholar] [CrossRef] [PubMed]
- Gelsinger, D.R.; DiRuggiero, J. Transcriptional landscape and regulatory roles of small noncoding RNAs in the oxidative stress response of the Haloarchaeon Haloferax volcanii. J. Bacteriol. 2018, 200, e00779-17. [Google Scholar] [CrossRef] [PubMed]
- Gunde-Cimerman, N.; Plemenitaš, A.; Oren, A. Strategies of adaptation of microorganisms of the three domains of life to high salt concentrations. FEMS Microbiol. Rev. 2018, 42, 353–375. [Google Scholar] [CrossRef]
- Oesterhelt, D.; Stoeckenius, W. Rhodopsin-like protein from the purple membrane of Halobacterium halobium. Nat. New Biol. 1971, 233, 149–152. [Google Scholar] [CrossRef] [PubMed]
- Oren, A.; Trüper, H. Anaerobic growth of halophilic archaeobacteria by reduction of dimethysulfoxide and trimethylamine N-oxide. FEMS Microbiol. Lett. 1990, 70, 33–36. [Google Scholar] [CrossRef]
- Ruepp, A.; Soppa, J. Fermentative arginine degradation in Halobacterium salinarium (Formerly Halobacterium halobium): Genes, gene products, and transcripts of the arcRACB gene cluster. J. Bacteriol. 1996, 178, 4942–4947. [Google Scholar] [CrossRef] [PubMed]
- Kaur, A.; Pan, M.; Meislin, M.; Facciotti, M.T.; El-Gewely, R.; Baliga, N.S. A systems view of haloarchaeal strategies to withstand stress from transition metals. Genome Res. 2006, 16, 841–854. [Google Scholar] [CrossRef]
- Coker, J.A.; DasSarma, P.; Kumar, J.; Müller, J.A.; DasSarma, S. Transcriptional profiling of the model Archaeon Halobacterium sp. NRC-1: Responses to changes in salinity and temperature. Saline Syst. 2007, 3, 6. [Google Scholar] [CrossRef]
- Baliga, N.S.; Bjork, S.J.; Bonneau, R.; Pan, M.; Iloanusi, C.; Kottemann, M.C.H.; Hood, L.; DiRuggiero, J. Systems level insights into the stress response to UV radiation in the halophilic archaeon Halobacterium NRC-1. Genome Res. 2004, 14, 1025–1035. [Google Scholar] [CrossRef] [PubMed]
- Bonneau, R.; Facciotti, M.T.; Reiss, D.J.; Schmid, A.K.; Pan, M.; Kaur, A.; Thorsson, V.; Shannon, P.; Johnson, M.H.; Bare, J.C.; et al. A predictive model for transcriptional control of physiology in a free living cell. Cell 2007, 131, 1354–1365. [Google Scholar] [CrossRef] [PubMed]
- Brooks, A.N.; Reiss, D.J.; Allard, A.; Wu, W.-J.; Salvanha, D.M.; Plaisier, C.L.; Chandrasekaran, S.; Pan, M.; Kaur, A.; Baliga, N.S. A system-level model for the microbial regulatory genome. Mol. Syst. Biol. 2014, 10, 740. [Google Scholar] [CrossRef]
- Koide, T.; Reiss, D.J.; Bare, J.C.; Pang, W.L.; Facciotti, M.T.; Schmid, A.K.; Pan, M.; Marzolf, B.; Van, P.T.; Lo, F.Y.; et al. Prevalence of transcription promoters within archaeal operons and coding sequences. Mol. Syst. Biol. 2009, 5, 285. [Google Scholar] [CrossRef]
- Ten-Caten, F.; Vêncio, R.Z.N.; Lorenzetti, A.P.R.; Zaramela, L.S.; Santana, A.C.; Koide, T. Internal RNAs overlapping coding sequences can drive the production of alternative proteins in archaea. RNA Biol. 2018, 15, 1119–1132. [Google Scholar] [CrossRef]
- Zaramela, L.S.; Vêncio, R.Z.N.; Ten-Caten, F.; Baliga, N.S.; Koide, T. Transcription start site associated RNAs (TSSaRNAs) are ubiquitous in all domains of life. PLoS ONE 2014, 9, e107680. [Google Scholar] [CrossRef]
- Gomes-Filho, J.V.; Zaramela, L.S.; da Silva Italiani, V.C.; Baliga, N.S.; Vêncio, R.Z.N.; Koide, T. Sense overlapping transcripts in IS1341-type transposase genes are functional non-coding RNAs in archaea. RNA Biol. 2015, 12, 490–500. [Google Scholar] [CrossRef]
- Stolt, P.; Zillig, W. Structure specific ds/ss-RNase activity in the extreme halophile Halobacterium salinarium. Nucleic Acids Res. 1993, 21, 5595–5599. [Google Scholar] [CrossRef]
- Wagner, E.G.H.; Simons, R.W. Antisense RNA control in bacteria, phages, and plasmids. Annu. Rev. Microbiol. 1994, 48, 713–742. [Google Scholar] [CrossRef]
- Leinonen, R.; Sugawara, H.; Shumway, M. The sequence read archive. Nucleic Acids Res. 2010, 39, D19–D21. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Kahles, A.; Behr, J.; Rätsch, G. MMR: A tool for read multi-mapper resolution. Bioinformatics 2015, 32, 770–772. [Google Scholar] [CrossRef]
- Quinlan, A.R. BEDTools: The Swiss-army tool for genome feature analysis. Curr. Protoc. Bioinform. 2014, 47, 11–12. [Google Scholar] [CrossRef]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative genomics viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef]
- Bare, J.C.; Koide, T.; Reiss, D.J.; Tenenbaum, D.; Baliga, N.S. Integration and visualization of systems biology data in context of the genome. BMC Bioinform. 2010, 11, 382. [Google Scholar] [CrossRef]
- Amman, F.; Wolfinger, M.T.; Lorenz, R.; Hofacker, I.L.; Stadler, P.F.; Findeiß, S. TSSAR: TSS annotation regime for dRNA-seq data. BMC Bioinform. 2014, 15, 89. [Google Scholar] [CrossRef]
- Pfeiffer, F.; Oesterhelt, D. A manual curation strategy to improve genome annotation: Application to a set of Haloarchael genomes. Life 2015, 5, 1427–1444. [Google Scholar] [CrossRef]
- Lorenz, R.; Bernhart, S.H.; Zu Siederdissen, C.H.; Tafer, H.; Flamm, C.; Stadler, P.F.; Hofacker, I.L. ViennaRNA Package 2.0. Algorithms Mol. Biol. 2011, 6, 26. [Google Scholar] [CrossRef]
- Ramírez, F.; Ryan, D.P.; Grüning, B.; Bhardwaj, V.; Kilpert, F.; Richter, A.S.; Heyne, S.; Dündar, F.; Manke, T. DeepTools2: A next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016, 44, W160–W165. [Google Scholar] [CrossRef]
- Crooks, G.E.; Hon, G.; Chandonia, J.M.; Brenner, S.E. WebLogo: A sequence logo generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef]
- Mi, H.; Muruganujan, A.; Casagrande, J.T.; Thomas, P.D. Large-scale gene function analysis with the PANTHER classification system. Nat. Protoc. 2013, 8, 1551–1566. [Google Scholar] [CrossRef]
- Dehal, P.S.; Joachimiak, M.P.; Price, M.N.; Bates, J.T.; Baumohl, J.K.; Chivian, D.; Friedland, G.D.; Huang, K.H.; Keller, K.; Novichkov, P.S.; et al. MicrobesOnline: An integrated portal for comparative and functional genomics. Nucleic Acids Res. 2009, 38, D396–D400. [Google Scholar] [CrossRef]
- Marchler-Bauer, A.; Bo, Y.; Han, L.; He, J.; Lanczycki, C.J.; Lu, S.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; et al. CDD/SPARCLE: Functional classification of proteins via subfamily domain architectures. Nucleic Acids Res. 2016, 45, D200–D203. [Google Scholar] [CrossRef]
- Xie, Y.; Wei, Y.; Shen, Y.; Li, X.; Zhou, H.; Tai, C.; Deng, Z.; Ou, H.-Y. TADB 2.0: An updated database of bacterial type II toxin-antitoxin loci. Nucleic Acids Res. 2018, 46, D749–D753. [Google Scholar] [CrossRef]
- Thomason, M.K.; Bischler, T.; Eisenbart, S.K.; Förstner, K.U.; Zhang, A.; Herbig, A.; Nieselt, K.; Sharma, C.M.; Storz, G. Global transcriptional start site mapping using differential RNA sequencing reveals novel antisense RNAs in Escherichia coli. J. Bacteriol. 2015, 197, 18–28. [Google Scholar] [CrossRef]
- Lybecker, M.; Zimmermann, B.; Bilusic, I.; Tukhtubaeva, N.; Schroeder, R. The double-stranded transcriptome of Escherichia coli. Proc. Natl. Acad. Sci. USA 2014, 111, 3134–3139. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq-A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, M.D.; Winsor, G.L.; Laird, M.R.; Brinkman, F.S.L. OrtholugeDB: A bacterial and archaeal orthology resource for improved comparative genomic analysis. Nucleic Acids Res. 2012, 41, D366–D376. [Google Scholar] [CrossRef] [PubMed]
- Babski, J.; Maier, L.-K.; Heyer, R.; Jaschinski, K.; Prasse, D.; Jäger, D.; Randau, L.; Schmitz, R.A.; Marchfelder, A.; Soppa, J. Small regulatory RNAs in Archaea. RNA Biol. 2014, 11, 484–493. [Google Scholar] [CrossRef]
- Kim, D.; Hong, J.S.-J.; Qiu, Y.; Nagarajan, H.; Seo, J.H.; Cho, B.K.; Tsai, S.F.; Palsson, B.Ø. Comparative analysis of regulatory elements between Escherichia coli and Klebsiella pneumoniae by genome-wide transcription start site profiling. PLoS Genet. 2012, 8, e1002867. [Google Scholar] [CrossRef]
- Wurtzel, O.; Sapra, R.; Chen, F.; Zhu, Y.; Simmons, B.A.; Sorek, R. A single-base resolution map of an archaeal transcriptome. Genome Res. 2010, 20, 133–141. [Google Scholar] [CrossRef]
- Carninci, P.; Sandelin, A.; Lenhard, B.; Katayama, S.; Shimokawa, K.; Ponjavic, J.; Semple, C.A.M.; Taylor, M.S.; Engström, P.G.; Frith, M.C.; et al. Genome-wide analysis of mammalian promoter architecture and evolution. Nat. Genet. 2006, 38, 626–635. [Google Scholar] [CrossRef]
- Bell, S.D.; Jackson, S.P. Mechanism and regulation of transcription in archaea. Curr. Opin. Microbiol. 2001, 4, 208–213. [Google Scholar] [CrossRef]
- Seitzer, P.; Wilbanks, E.G.; Larsen, D.J.; Facciotti, M.T. A Monte Carlo-based framework enhances the discovery and interpretation of regulatory sequence motifs. BMC Bioinform. 2012, 13, 317. [Google Scholar] [CrossRef]
- Dar, D.; Prasse, D.; Schmitz, R.A.; Sorek, R. Widespread formation of alternative 3′ UTR isoforms via transcription termination in archaea. Nat. Microbiol. 2016, 1, 16143. [Google Scholar] [CrossRef]
- Brenneis, M.; Hering, O.; Lange, C.; Soppa, J. Experimental characterization of cis-acting elements important for translation and transcription in halophilic archaea. PLoS Genet. 2007, 3, e229. [Google Scholar] [CrossRef]
- Bouvier, M.; Sharma, C.M.; Mika, F.; Nierhaus, K.H.; Vogel, J. Small RNA binding to 5′ mRNA coding region inhibits translational initiation. Mol. Cell 2008, 32, 827–837. [Google Scholar] [CrossRef]
- Fozo, E.M.; Hemm, M.R.; Storz, G. Small toxic proteins and the antisense RNAs that repress them. Microbiol. Mol. Biol. Rev. 2008, 72, 579–589. [Google Scholar] [CrossRef]
- Pfeifer, F.; Krüger, K.; Röder, R.; Mayr, A.; Ziesche, S.; Offner, S. Gas vesicle formation in halophilic Archaea. Arch. Microbiol. 1997, 167, 259–268. [Google Scholar] [CrossRef]
- Csiszàr, K.; Houmard, J.; Damerval, T.; de Marsac, N.T. Transcriptional analysis of the cyanobacterial gvpABC operon in differentiated cells: Occurrence of an antisense RNA complementary to three overlapping transcripts. Gene 1987, 60, 29–37. [Google Scholar] [CrossRef]
- Tarasov, V.; Schwaiger, R.; Furtwängler, K.; Dyall-Smith, M.; Oesterhelt, D. A small basic protein from the brz-brb operon is involved in regulation of bop transcription in Halobacterium salinarum. BMC Mol. Biol. 2011, 12, 42. [Google Scholar] [CrossRef]
- Harms, A.; Brodersen, D.E.; Mitarai, N.; Gerdes, K. Toxins, targets, and triggers: An overview of toxin-antitoxin biology. Mol. Cell 2018, 70, 768–784. [Google Scholar] [CrossRef]
- Tang, T.H.; Polacek, N.; Zywicki, M.; Huber, H.; Brugger, K.; Garrett, R.; Bachellerie, J.P.; Hüttenhofer, A. Identification of novel non-coding RNAs as potential antisense regulators in the archaeon Sulfolobus solfataricus. Mol. Microbiol. 2005, 55, 469–481. [Google Scholar] [CrossRef]
- Ellis, M.J.; Trussler, R.S.; Haniford, D.B. A cis-encoded sRNA, Hfq and mRNA secondary structure act independently to suppress IS200 transposition. Nucleic Acids Res. 2015, 43, 6511–6527. [Google Scholar] [CrossRef]
- Pircher, A.; Gebetsberger, J.; Polacek, N. Ribosome-associated ncRNAs: An emerging class of translation regulators. RNA Biol. 2014, 11, 1335–1339. [Google Scholar] [CrossRef]
- Wyss, L.; Waser, M.; Gebetsberger, J.; Zywicki, M.; Polacek, N. mRNA-specific translation regulation by a ribosome-associated ncRNA in Haloferax volcanii. Sci. Rep. 2018, 8, 12502. [Google Scholar] [CrossRef]
- Raghavan, R.; Sloan, D.B.; Ochman, H.C. Antisense transcription is pervasive but rarely conserved in enteric bacteria. MBio 2012, 3, e00156-12. [Google Scholar] [CrossRef]
- Dugar, G.; Herbig, A.; Förstner, K.U.; Heidrich, N.; Reinhardt, R.; Nieselt, K.; Sharma, C.M. High-resolution transcriptome maps reveal strain-specific regulatory features of multiple Campylobacter jejuni isolates. PLoS Genet. 2013, 9, e1003495. [Google Scholar] [CrossRef]
- Shao, W.; Price, M.N.; Deutschbauer, A.M.; Romine, M.F.; Arkin, A.P. Conservation of transcription start sites within genes across a bacterial genus. MBio 2014, 5, e01398-14. [Google Scholar] [CrossRef]
- Kopf, M.; Klähn, S.; Scholz, I.; Hess, W.R.; Voß, B. Variations in the non-coding transcriptome as a driver of inter-strain divergence and physiological adaptation in bacteria. Sci. Rep. 2015, 5, 9560. [Google Scholar] [CrossRef]
- Mei, Y.; Liu, H.; Zhang, S.; Yang, M.; Hu, C.; Zhang, J.; Shen, P.; Chen, X. Effects of salinity on the cellular physiological responses of Natrinema sp. J7-2. PLoS ONE 2017, 12, e0184974. [Google Scholar] [CrossRef]
- Irnov, I.; Sharma, C.M.; Vogel, J.; Winkler, W.C. Identification of regulatory RNAs in Bacillus subtilis. Nucleic Acids Res. 2010, 38, 6637–6651. [Google Scholar] [CrossRef]
- Van der Meulen, S.B.; de Jong, A.; Kok, J. Transcriptome landscape of Lactococcus lactis reveals many novel RNAs including a small regulatory RNA involved in carbon uptake and metabolism. RNA Biol. 2016, 13, 353–366. [Google Scholar] [CrossRef]
- Garber, M.; Grabherr, M.G.; Guttman, M.; Trapnell, C. Computational methods for transcriptome annotation and quantification using RNA-seq. Nat. Methods 2011, 8, 469. [Google Scholar] [CrossRef]
- Santangelo, T.J.; Reeve, J.N. Archaeal RNA polymerase is sensitive to intrinsic termination directed by transcribed and remote sequences. J. Mol. Biol. 2006, 355, 196–210. [Google Scholar] [CrossRef] [PubMed]
- Garalde, D.R.; Snell, E.A.; Jachimowicz, D.; Sipos, B.; Lloyd, J.H.; Bruce, M.; Pantic, N.; Admassu, T.; James, P.; Warland, A.; et al. Highly parallel direct RNA sequencing on an array of nanopores. Nat. Methods 2018, 15, 201–206. [Google Scholar] [CrossRef]
- Brosius, J.; Gould, S.J. On “genomenclature”: A comprehensive (and respectful) taxonomy for pseudogenes and other “junk DNA”. Proc. Natl. Acad. Sci. USA 1992, 89, 10706–10710. [Google Scholar] [CrossRef]
- Goyal, A.; Fiškin, E.; Gutschner, T.; Polycarpou-Schwarz, M.; Groß, M.; Neugebauer, J.; Gandhi, M.; Caudron-Herger, M.; Benes, V.; Diederichs, S. A cautionary tale of sense-antisense gene pairs: Independent regulation despite inverse correlation of expression. Nucleic Acids Res. 2017, 45, 12496–12508. [Google Scholar] [CrossRef]
- Vogel, C.; Marcotte, E.M. Insights into regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet. 2012, 13, 227–232. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| aTSS ID | asRNA ID | Strand | Start | End | Size (nt) | Locus | Annotation |
|---|---|---|---|---|---|---|---|
| aTSS_1555 | VNG_as12280_1555 | + | 16743 | 16863 | 120 | VNG_RS12280 | gvpL |
| aTSS_1556 | VNG_as12280_1556 | + | 17055 | 17138 | 83 | VNG_RS12280 | gvpL |
| aTSS_1557 | VNG_as12290_1557 | + | 18092 | 18168 | 76 | VNG_RS12290 | gvpJ |
| aTSS_1558 | VNG_as13760_1558 | + | 18390 | 18428 | 38 | VNG_RS13760 | gvpI |
| aTSS_1559 | VNG_as13760_1559 | + | 18615 | 18643 | 28 | VNG_RS13760 | gvpI |
| aTSS_1560 | VNG_as12295_1560 | + | 18698 | 18722 | 24 | VNG_RS12295 | gvpH |
| aTSS_1565 | VNG_as12315_1565 | + | 21000 | 21165 | 165 | VNG_RS12315 | gvpD |
| aTSS_1567 | VNG_as12315_1567 | + | 22084 | 22106 | 22 | VNG_RS12315 | gvpD |
| aTSS_1568 | VNG_as12315_1568 | + | 22128 | 22292 | 164 | VNG_RS12315 | gvpD |
| aTSS_1569 | VNG_as12325_1569 | − | 22865 | 22964 | 99 | VNG_RS12325 | gvpC |
| aTSS_1570 | VNG_as12325_1570 | − | 23888 | 23940 | 52 | VNG_RS12325 | gvpC |
| aTSS ID | asRNA ID | Strand | Start | End | Size (nt) | Locus | Annotation |
|---|---|---|---|---|---|---|---|
| aTSS_175 | VNG_as00745_175 | + | 155806 | 155906 | 100 | VNG_RS00745 | halorhodopsin |
| *daTSS_36 | VNG_da3105F_36 | − | 1088797 | 1089100 | 303 | VNG_RS05710 VNG_OE3105F | brz—bacteriorhodopsin regulating zinc finger protein; brb—bacteriorhodopsin-regulating basic protein |
| aTSS_824 | VNG_as05715_824 | − | 1089545 | 1089615 | 70 | VNG_RS05715 | bacteriorhodopsin |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Almeida, J.P.P.; Vêncio, R.Z.N.; Lorenzetti, A.P.R.; ten-Caten, F.; Gomes-Filho, J.V.; Koide, T. The Primary Antisense Transcriptome of Halobacterium salinarum NRC-1. Genes 2019, 10, 280. https://doi.org/10.3390/genes10040280
de Almeida JPP, Vêncio RZN, Lorenzetti APR, ten-Caten F, Gomes-Filho JV, Koide T. The Primary Antisense Transcriptome of Halobacterium salinarum NRC-1. Genes. 2019; 10(4):280. https://doi.org/10.3390/genes10040280
Chicago/Turabian Stylede Almeida, João Paulo Pereira, Ricardo Z. N. Vêncio, Alan P. R. Lorenzetti, Felipe ten-Caten, José Vicente Gomes-Filho, and Tie Koide. 2019. "The Primary Antisense Transcriptome of Halobacterium salinarum NRC-1" Genes 10, no. 4: 280. https://doi.org/10.3390/genes10040280
APA Stylede Almeida, J. P. P., Vêncio, R. Z. N., Lorenzetti, A. P. R., ten-Caten, F., Gomes-Filho, J. V., & Koide, T. (2019). The Primary Antisense Transcriptome of Halobacterium salinarum NRC-1. Genes, 10(4), 280. https://doi.org/10.3390/genes10040280