Three New Integration Vectors and Fluorescent Proteins for Use in the Opportunistic Human Pathogen Streptococcus pneumoniae


Abstract
:1. Introduction
2. Materials and Methods
2.1. Growth Conditions
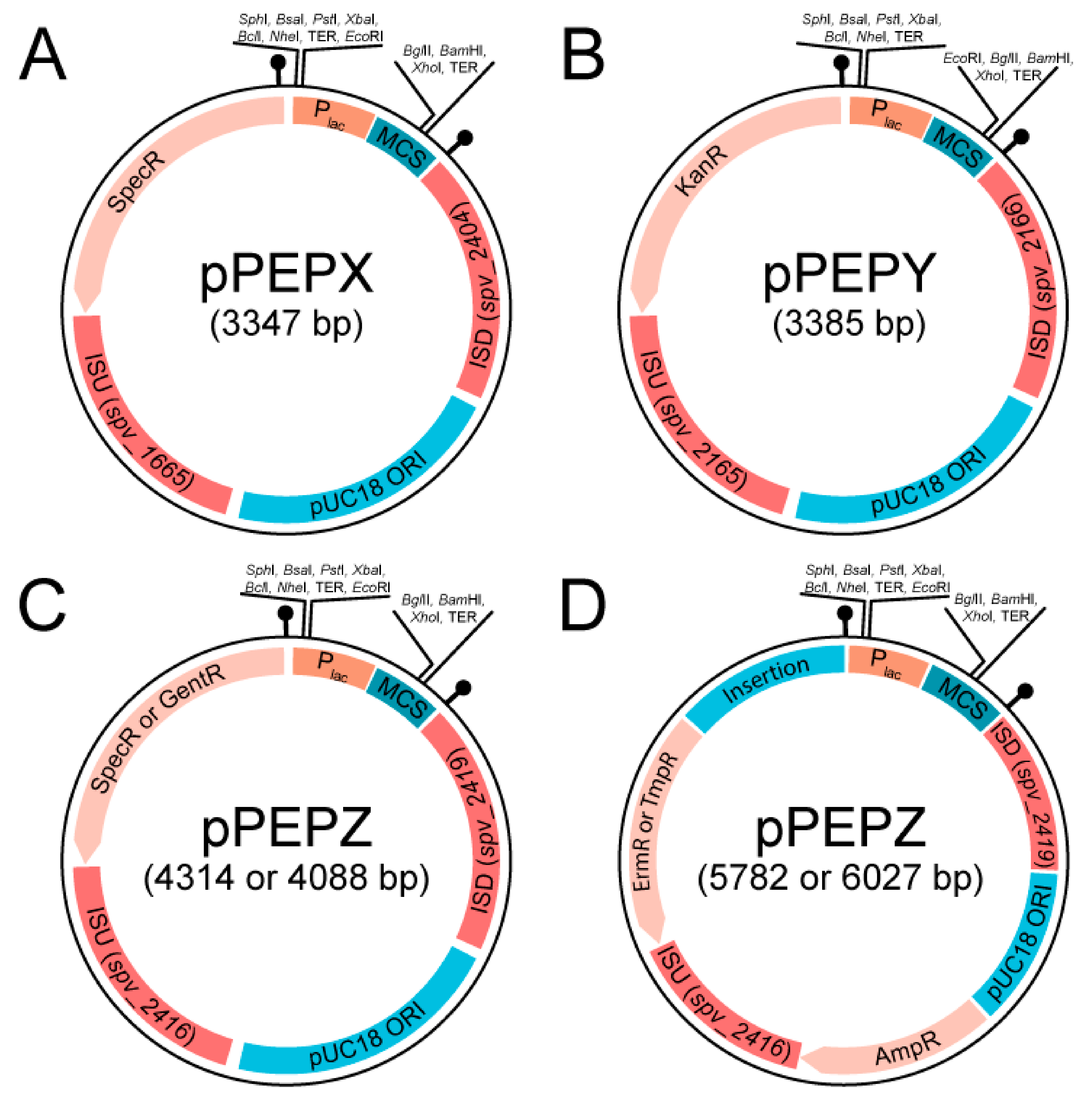
2.2. Plasmid Construction
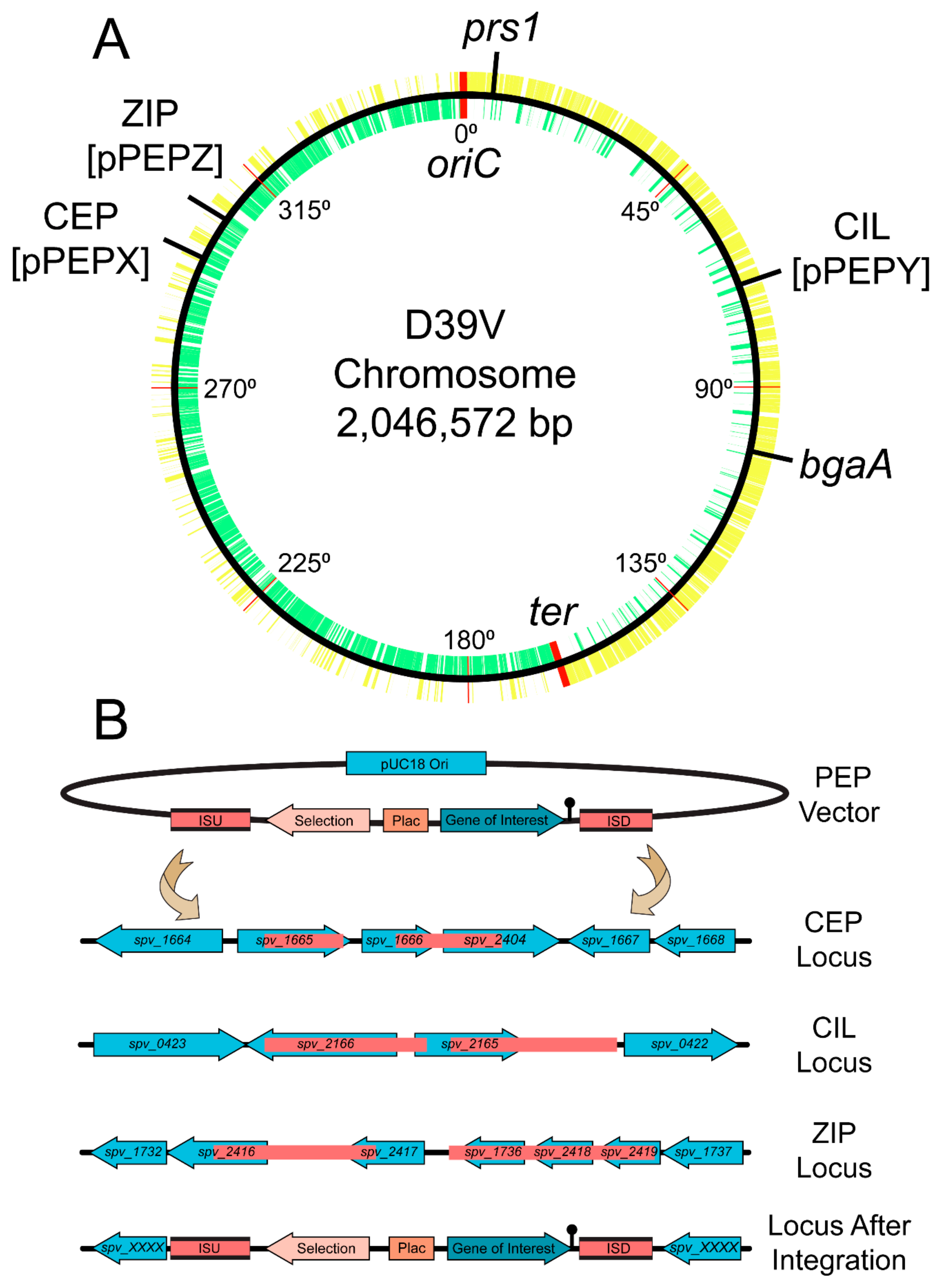
2.3. Strain Construction
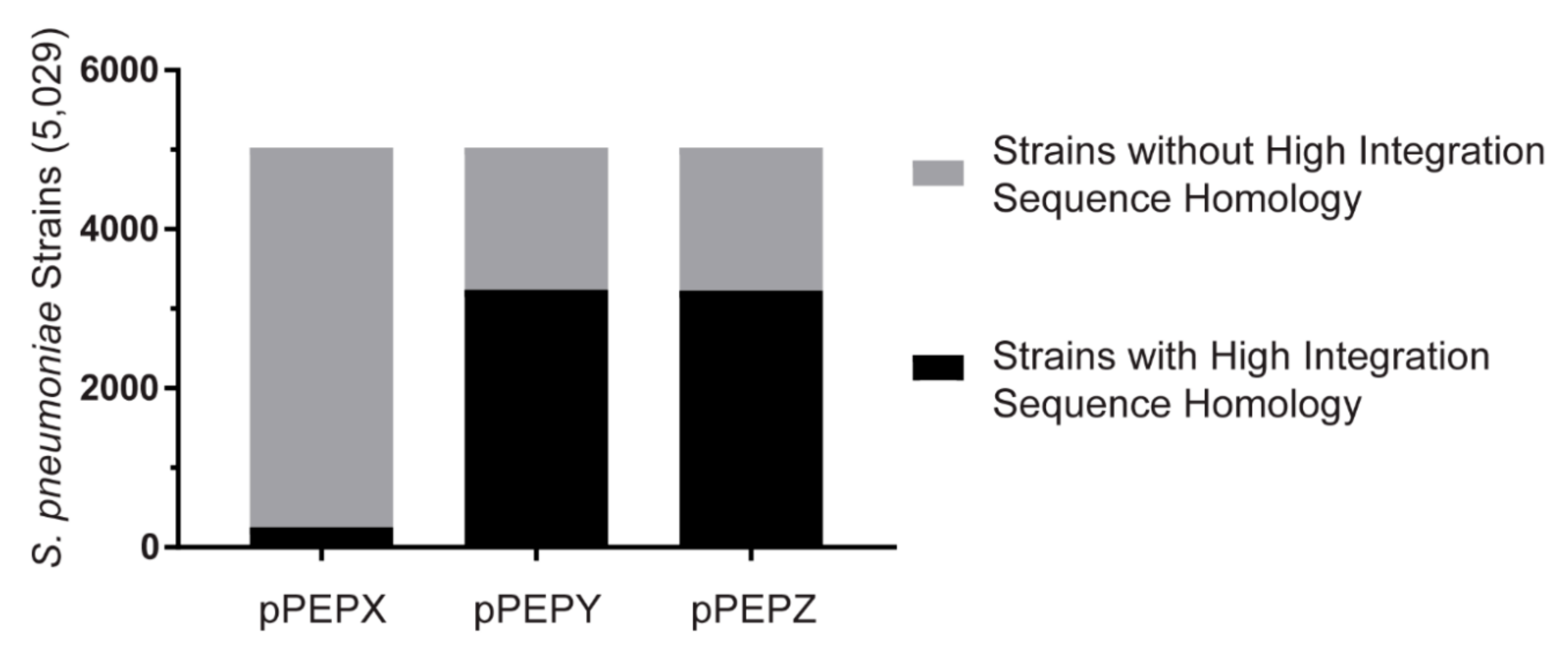
2.4. Database BLAST
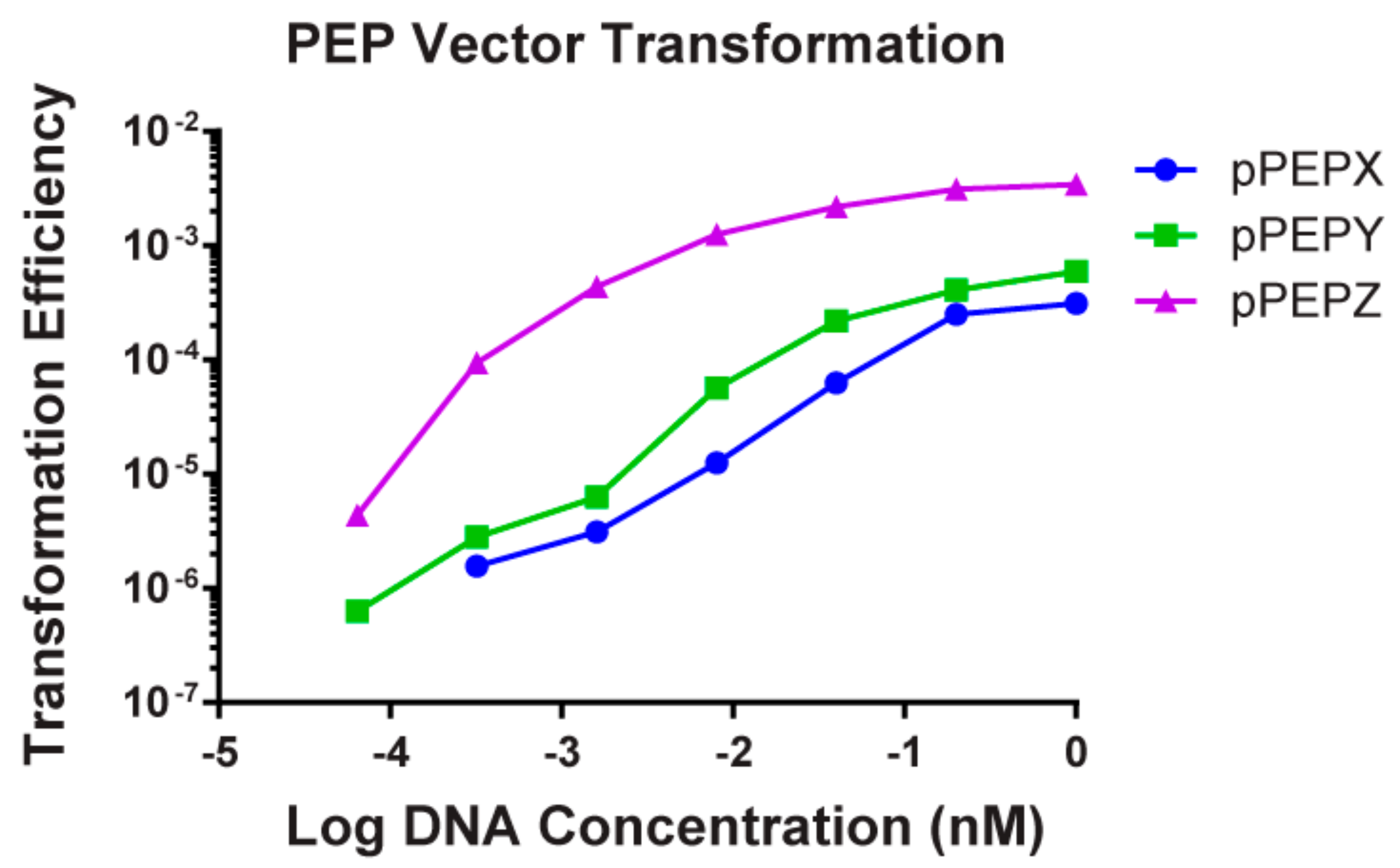
2.5. Transformation Assay
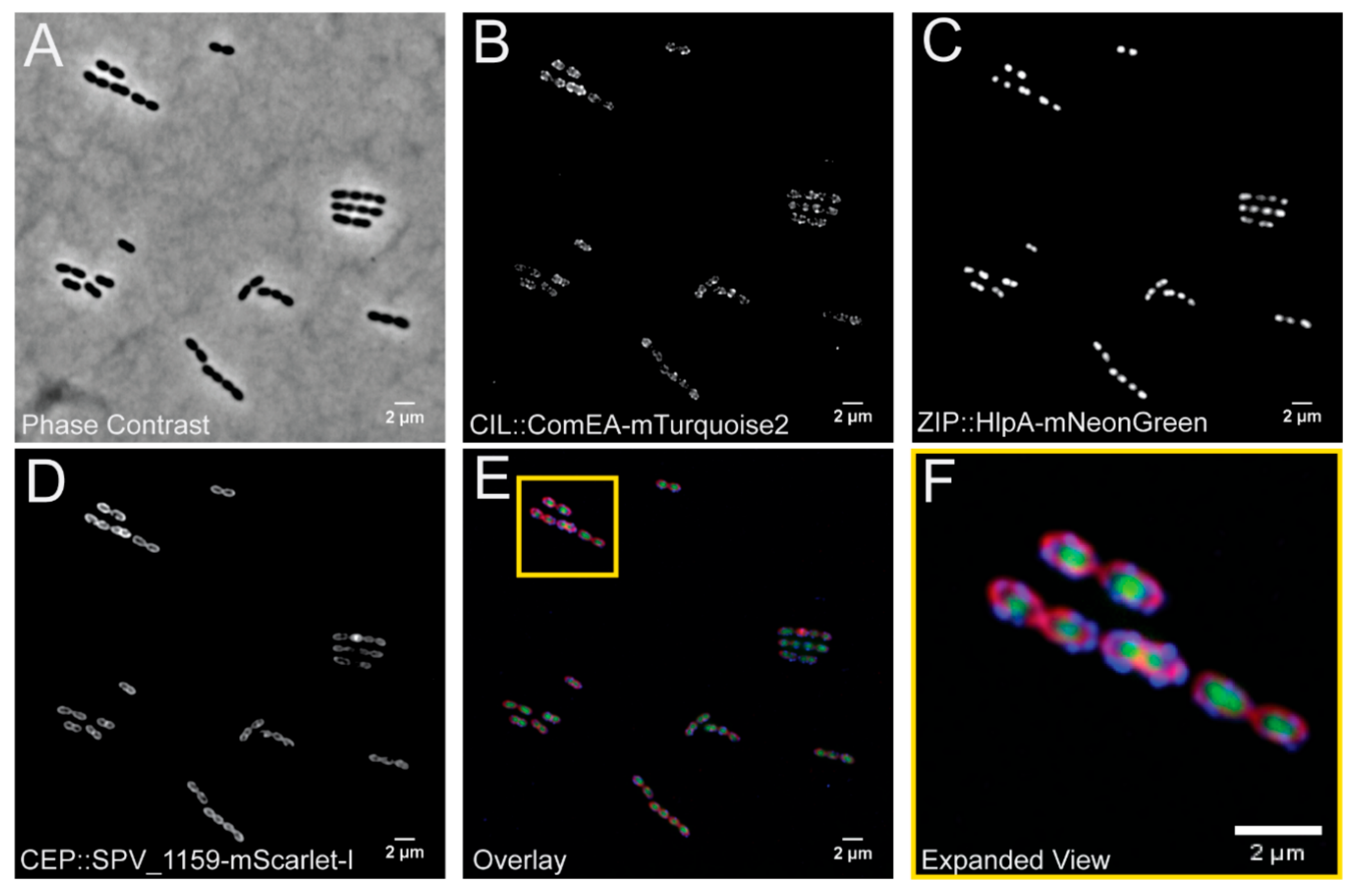
2.6. Microscopy
3. Results
3.1. Design and Construction of pPEPX, pPEPY and pPEPZ
3.2. High Transformation Efficiency of pPEPX, pPEPY and pPEPZ
3.3. Simultaneous Integration and Expression of Three New Fluorescent Proteins for S. pneumoniae from the pPEPX, pPEPY and pPEPZ Loci
3.4. Broad Pneumococcal Strain Range for pPEPY and pPEPZ
3.5. Efficient Cloning in Multidrug Resistant Clinical Isolates Using PEP Vectors
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Bryce, J.; Boschi-Pinto, C.; Shibuya, K.; Black, R.E. WHO estimates of the causes of death in children. Lancet 2005, 365, 1147–1152. [Google Scholar] [CrossRef]
- Khalil, A.S.; Collins, J.J. Synthetic biology: Applications come of age. Nat. Rev. Genet. 2010, 11, 367–379. [Google Scholar] [CrossRef] [PubMed]
- Troeger, C.; Blacker, B.; Khalil, I.A.; Rao, P.C.; Cao, J.; Zimsen, S.R.M.; Albertson, S.B.; Deshpande, A.; Farag, T.; Abebe, Z.; et al. Estimates of the global, regional, and national morbidity, mortality, and aetiologies of lower respiratory infections in 195 countries, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Infect. Dis. 2018, 18, 1191–1210. [Google Scholar] [CrossRef]
- Robertson, G.T.; Ng, W.-L.; Foley, J.; Gilmour, R.; Winkler, M.E. Global transcriptional analysis of clpP mutations of type 2 Streptococcus pneumoniae and their effects on physiology and virulence. J. Bacteriol. 2002, 184, 3508–3520. [Google Scholar] [PubMed]
- Halfmann, A.; Hakenbeck, R.; Brückner, R. A new integrative reporter plasmid for Streptococcus pneumoniae. FEMS Microbiol. Lett. 2007, 268, 217–224. [Google Scholar]
- Eberhardt, A.; Wu, L.J.; Errington, J.; Vollmer, W.; Veening, J.-W. Cellular localization of choline-utilization proteins in Streptococcus pneumoniae using novel fluorescent reporter systems. Mol. Microbiol. 2009, 74, 395–408. [Google Scholar] [CrossRef] [PubMed]
- Overkamp, W.; Beilharz, K.; Detert Oude Weme, R.; Solopova, A.; Karsens, H.; Kovács, Á.T.; Kok, J.; Kuipers, O.P.; Veening, J.-W. Benchmarking various green fluorescent protein variants in bacillus subtilis, Streptococcus pneumoniae, and Lactococcus lactis for live cell imaging. Appl. Environ. Microbiol. 2013, 79, 6481–6490. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.; Grau, M.A.; Singh, A.K.; Woodiga, S.A.; King, S.J. The role of neuraminidase-producing bacteria in exposing cryptic carbohydrate receptors for Streptococcus gordonii adherence. Infect. Immun. 2018, 86, e00068–18. [Google Scholar] [CrossRef]
- Dalia, A.B.; Standish, A.J.; Weiser, J.N. Three surface exoglycosidases from Streptococcus pneumoniae, NanA, BgaA, and StrH, promote resistance to opsonophagocytic killing by human neutrophils. Infect. Immun. 2010, 78, 2108–2116. [Google Scholar] [CrossRef]
- King, S.J.; Hippe, K.R.; Weiser, J.N. Deglycosylation of human glycoconjugates by the sequential activities of exoglycosidases expressed by Streptococcus pneumoniae. Mol. Microbiol. 2006, 59, 961–974. [Google Scholar] [CrossRef]
- Guiral, S.; Henard, V.; Laaberki, M.-H.; Granadel, C.; Prudhomme, M.; Martin, B.; Claverys, J.-P. Construction and evaluation of a chromosomal expression platform (CEP) for ectopic, maltose-driven gene expression in Streptococcus pneumoniae. Microbiology 2006, 152, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Sorg, R.A.; Kuipers, O.P.; Veening, J.-W. Gene expression platform for synthetic biology in the human pathogen Streptococcus pneumoniae. ACS Synth. Biol. 2014, 4, 228–239. [Google Scholar] [CrossRef]
- Aprianto, R.; Slager, J.; Holsappel, S.; Veening, J.-W. High-resolution analysis of the pneumococcal transcriptome under a wide range of infection-relevant conditions. Nucleic Acids Res. 2018, 46, 9990–10006. [Google Scholar] [CrossRef]
- Slager, J.; Aprianto, R.; Veening, J.-W. Deep genome annotation of the opportunistic human pathogen Streptococcus pneumoniae D39. Nucleic Acids Res. 2018, 46, 9971–9989. [Google Scholar] [CrossRef] [PubMed]
- Domenech, A.; Slager, J.; Veening, J.-W. Antibiotic-induced cell chaining triggers pneumococcal competence by reshaping quorum sensing to autocrine-like signaling. Cell Rep. 2018, 25, 2390–2400. [Google Scholar] [CrossRef]
- Martin, B.; Garcia, P.; Castanié, M.; Claverys, J. The recA gene of Streptococcus pneumoniae is part of a competence-induced operon and controls lysogenic induction. Mol. Microbiol. 1995, 15, 367–379. [Google Scholar] [CrossRef]
- Slager, J.; Kjos, M.; Attaiech, L.; Veening, J.-W. Antibiotic-induced replication stress triggers bacterial competence by increasing gene dosage near the origin. Cell 2014, 157, 395–406. [Google Scholar] [CrossRef]
- Sorg, R.A. Engineering Approaches to Investigate Pneumococcal Gene Expression Regulation and Antibiotic Resistance Development; Rijksuniversiteit: Groningen, The Netherlands, 2016; ISBN 9036792584. [Google Scholar]
- Fischer, K.E.; Bremer, E. Activity of the osmotically regulated yqiHIK promoter from Bacillus subtilis is controlled at a distance. J. Bacteriol. 2012, 194, 5197–5208. [Google Scholar] [CrossRef]
- Liu, X.; Gallay, C.; Kjos, M.; Domenech, A.; Slager, J.; van Kessel, S.P.; Knoops, K.; Sorg, R.A.; Zhang, J.; Veening, J. High-throughput CRISPRi phenotyping identifies new essential genes in Streptococcus pneumoniae. Mol. Syst. Biol. 2017, 13, 931. [Google Scholar] [CrossRef] [PubMed]
- Minnen, A.; Attaiech, L.; Thon, M.; Gruber, S.; Veening, J. SMC is recruited to oriC by ParB and promotes chromosome segregation in Streptococcus pneumoniae. Mol. Microbiol. 2011, 81, 676–688. [Google Scholar] [CrossRef]
- Goedhart, J.; von Stetten, D.; Noirclerc-Savoye, M.; Lelimousin, M.; Joosen, L.; Hink, M.A.; van Weeren, L.; Gadella, T.W.J. Jr.; Royant, A. Structure-guided evolution of cyan fluorescent proteins towards a quantum yield of 93%. Nat. Commun. 2012, 3, 751. [Google Scholar] [CrossRef]
- Shaner, N.C.; Lambert, G.G.; Chammas, A.; Ni, Y.; Cranfill, P.J.; Baird, M.A.; Sell, B.R.; Allen, J.R.; Day, R.N.; Israelsson, M. A bright monomeric green fluorescent protein derived from Branchiostoma lanceolatum. Nat. Methods 2013, 10, 407–409. [Google Scholar] [CrossRef]
- Bindels, D.S.; Haarbosch, L.; van Weeren, L.; Postma, M.; Wiese, K.E.; Mastop, M.; Aumonier, S.; Gotthard, G.; Royant, A.; Hink, M.A.; et al. mScarlet: A bright monomeric red fluorescent protein for cellular imaging. Nat. Methods 2016, 14, 53–56. [Google Scholar] [CrossRef] [PubMed]
- Ohtsubo, Y.; Ikeda-Ohtsubo, W.; Nagata, Y.; Tsuda, M. GenomeMatcher: A graphical user interface for DNA sequence comparison. BMC Bioinform. 2008, 9, 376. [Google Scholar] [CrossRef]
- Rasband, W.S. ImageJ. US National Institutes of Health; Bethesda: Rockville, MD, USA, 1997. [Google Scholar]
- Anderson, J.; Dueber, J.E.; Leguia, M.; Wu, G.C.; Goler, J.A.; Arkin, A.P.; Keasling, J.D. BglBricks: A flexible standard for biological part assembly. J. Biol. Eng. 2010, 4, 1. [Google Scholar] [CrossRef]
- Lau, P.C.Y.; Sung, C.K.; Lee, J.H.; Morrison, D.A.; Cvitkovitch, D.G. PCR ligation mutagenesis in transformable streptococci: Application and efficiency. J. Microbiol. Methods 2002, 49, 193–205. [Google Scholar] [CrossRef]
- Lee, M.S.; Seok, C.; Morrison, D.A. Insertion-duplication mutagenesis in Streptococcus pneumoniae: Targeting fragment length is a critical parameter in use as a random insertion tool. Appl. Environ. Microbiol. 1998, 64, 4796–4802. [Google Scholar] [PubMed]
- Beilharz, K.; van Raaphorst, R.; Kjos, M.; Veening, J.-W. Red fluorescent proteins for gene expression and protein localization studies in Streptococcus pneumoniae and efficient transformation with Gibson assembled DNA. Appl. Environ. Microbiol. 2015, 81, 7244–7252. [Google Scholar] [CrossRef] [PubMed]
- Bergé, M.J.; Kamgoué, A.; Martin, B.; Polard, P.; Campo, N.; Claverys, J.-P. Midcell recruitment of the DNA uptake and virulence nuclease, EndA, for pneumococcal transformation. PLoS Pathog. 2013, 9, e1003596. [Google Scholar] [CrossRef]
- Kjos, M.; Aprianto, R.; Fernandes, V.E.; Andrew, P.W.; van Strijp, J.A.G.; Nijland, R.; Veening, J.-W. Bright fluorescent Streptococcus pneumoniae for live-cell imaging of host-pathogen interactions. J. Bacteriol. 2015, 197, 807–818. [Google Scholar] [CrossRef]
- Kjos, M.; Veening, J. Tracking of chromosome dynamics in live Streptococcus pneumoniae reveals that transcription promotes chromosome segregation. Mol. Microbiol. 2014, 91, 1088–1105. [Google Scholar] [CrossRef] [PubMed]
- Aprianto, R.; Slager, J.; Holsappel, S.; Veening, J.-W. Time-resolved dual RNA-seq reveals extensive rewiring of lung epithelial and pneumococcal transcriptomes during early infection. Genome Biol. 2016, 17, 198. [Google Scholar] [CrossRef] [PubMed]
- Chewapreecha, C.; Harris, S.R.; Croucher, N.J.; Turner, C.; Marttinen, P.; Cheng, L.; Pessia, A.; Aanensen, D.M.; Mather, A.E.; Page, A.J. Dense genomic sampling identifies highways of pneumococcal recombination. Nat. Genet. 2014, 46, 305–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, X.-Y.; Zhi, X.-Y.; Li, H.-W.; Klenk, H.-P.; Li, W.-J. Comparative genomics of the bacterial genus Streptococcus illuminates evolutionary implications of species groups. PLoS ONE 2014, 9, e101229. [Google Scholar] [CrossRef] [PubMed]
- Evans, B.A.; Rozen, D.E. Significant variation in transformation frequency in Streptococcus pneumoniae. ISME J. 2013, 7, 791–799. [Google Scholar] [CrossRef] [PubMed]
- McGee, L.; McDougal, L.; Zhou, J.; Spratt, B.G.; Tenover, F.C.; George, R.; Hakenbeck, R.; Hryniewicz, W.; Lefevre, J.C.; Tomasz, A. Nomenclature of major antimicrobial-resistant clones of Streptococcus pneumoniae defined by the pneumococcal molecular epidemiology network. J. Clin. Microbiol. 2001, 39, 2565–2571. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Plasmid Name | Features | Selection-E. coli | Selection-S. pneumoniae |
|---|---|---|---|
| pPEPX | Promoterless integrative plasmid with terminator | Spec 50 µg/mL | Spec 200 µg/mL |
| pPEPX-Plac | Integrative plasmid with IPTG inducible promoter | Spec 50 µg/mL | Spec 200 µg/mL |
| pPEPY | Promoterless integrative plasmid with terminator | Kan 50 µg/mL | Kan 250 µg/mL |
| pPEPY-Plac | Integrative plasmid with IPTG inducible promoter | Kan 50 µg/mL | Kan 250 µg/mL |
| pASR100 (pPEPZ-Plac) | Integrative plasmid with IPTG inducible promoter | Spec 50 µg/mL | Spec 200 µg/mL |
| pASR102 (pPEPZ-Plac) | Integrative plasmid with IPTG inducible promoter | Gent 40 µg/mL | Gent 50 µg/mL |
| pASR103 (pPEPZ-Plac) | Integrative plasmid with IPTG inducible promoter (Amp selection for E. coli) | Amp 100 µg/mL | Erm 0.5 µg/mL |
| pASR130 (pPEPZ-Plac) | Integrative plasmid with IPTG inducible promoter (Amp selection for E. coli) | Amp 100 µg/mL | Tmp 10 µg/mL |
| pPEPY-Plac-Link-mTurquoise2 | Integrative plasmid with IPTG inducible promoter and linker for C-terminal fusion of mTurquoise2 | Kan 50 µg/mL | Kan 250 µg/mL |
| pPEPY-Plac-Link-mNeonGreen | Integrative plasmid with IPTG inducible promoter and linker for C-terminal fusion of mNeonGreen | Kan 50 µg/mL | Kan 250 µg/mL |
| pPEPY-Plac-Link-mScarlet-I | Integrative plasmid with IPTG inducible promoter and linker for C-terminal fusion of mScarlet-I | Kan 50 µg/mL | Kan 250 µg/mL |
| Strain | Serotype | Susceptibility | PEP Transformable Based on Sequence | Transformation Efficiency (pPEPX) | Transformation Efficiency (pPEPY) | Transformation Efficiency (pPEPZ *) |
|---|---|---|---|---|---|---|
| PMEN1 | 23F | Erm, Spec, Kan | pPEPY and pPEPZ | 2.56 × 10−8 | 2.61 × 10−7 | 8.17 × 10−6 |
| PMEN3 | 9V | Erm, Spec, Kan | pPEPZ | 2.49 × 10−8 | 3.31 × 10−7 | 2.26 × 10−8 |
| PMEN14 | 19F | Gent, Spec, Kan | pPEPZ | 8.10 × 10−7 | 8.52 × 10−7 | 1.43 × 10−6 |
| PMEN18 | 14 | Gent, Spec, Kan | pPEPY and pPEPZ | 3.77 × 10−7 | 0 | 2.16 × 10−6 |
| PMEN28 | 1 | Erm, Gent, Spec, Kan | N/D | 8.79 × 10−8 | 7.95 × 10−8 | 1.03 × 10−6 |
| TIGR4 | 4 | Erm, Gent, Spec, Kan, Tmp | pPEPX and pPEPZ | 4.98 × 10−6 | 0 | 2.42 × 10−5 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Keller, L.E.; Rueff, A.-S.; Kurushima, J.; Veening, J.-W. Three New Integration Vectors and Fluorescent Proteins for Use in the Opportunistic Human Pathogen Streptococcus pneumoniae. Genes 2019, 10, 394. https://doi.org/10.3390/genes10050394
Keller LE, Rueff A-S, Kurushima J, Veening J-W. Three New Integration Vectors and Fluorescent Proteins for Use in the Opportunistic Human Pathogen Streptococcus pneumoniae. Genes. 2019; 10(5):394. https://doi.org/10.3390/genes10050394
Chicago/Turabian StyleKeller, Lance E., Anne-Stéphanie Rueff, Jun Kurushima, and Jan-Willem Veening. 2019. "Three New Integration Vectors and Fluorescent Proteins for Use in the Opportunistic Human Pathogen Streptococcus pneumoniae" Genes 10, no. 5: 394. https://doi.org/10.3390/genes10050394
APA StyleKeller, L. E., Rueff, A. -S., Kurushima, J., & Veening, J. -W. (2019). Three New Integration Vectors and Fluorescent Proteins for Use in the Opportunistic Human Pathogen Streptococcus pneumoniae. Genes, 10(5), 394. https://doi.org/10.3390/genes10050394