Illegitimate and Repeated Genomic Integration of Cell-Free Chromatin in the Aetiology of Somatic Mosaicism, Ageing, Chronic Diseases and Cancer

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Cell-Free Chromatin (cfCh): Background
2.1. Cell-Free Chromatin Versus Cell-Free DNA
2.2. Uptake by Healthy Cells of cfCh Released into Circulation
2.3. Uptake by Healthy Cells of cfCh Released Locally from Dying Cells
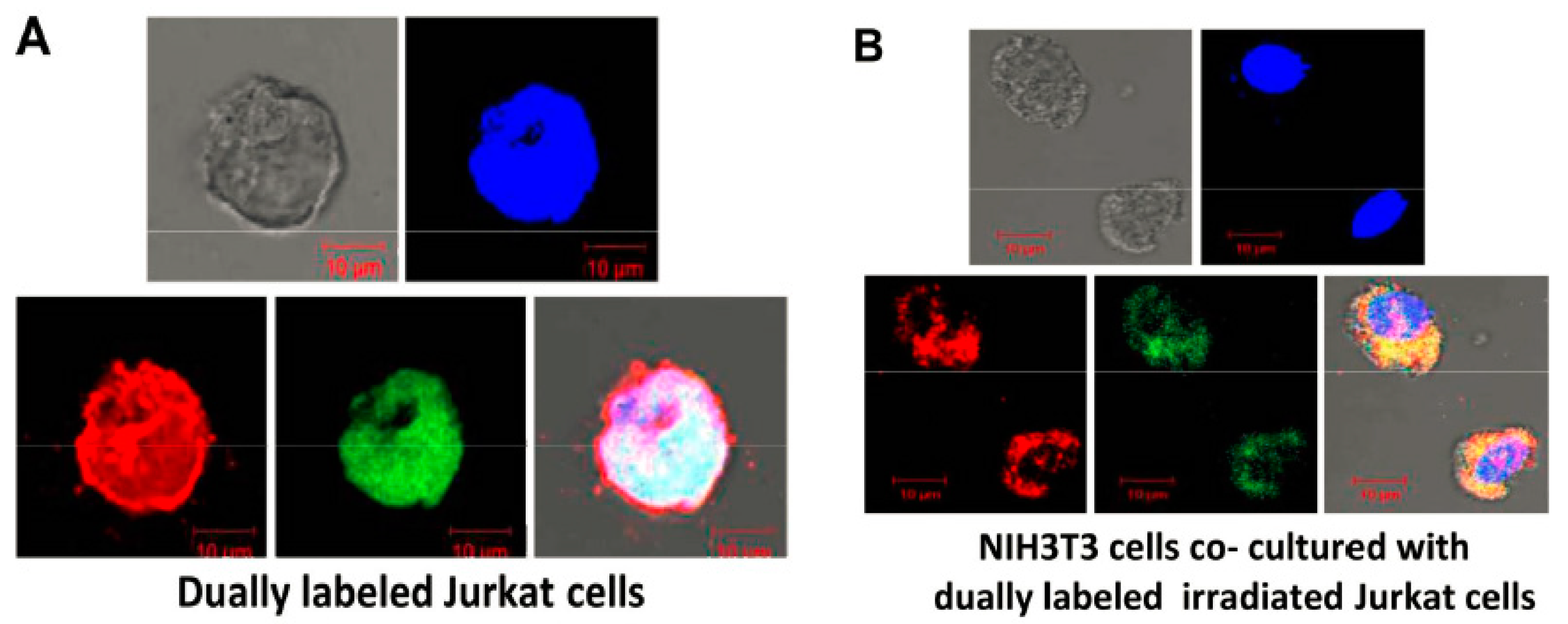
2.4. Uptake of cfCh Released from Circulating Tumour Cells at Target Sites
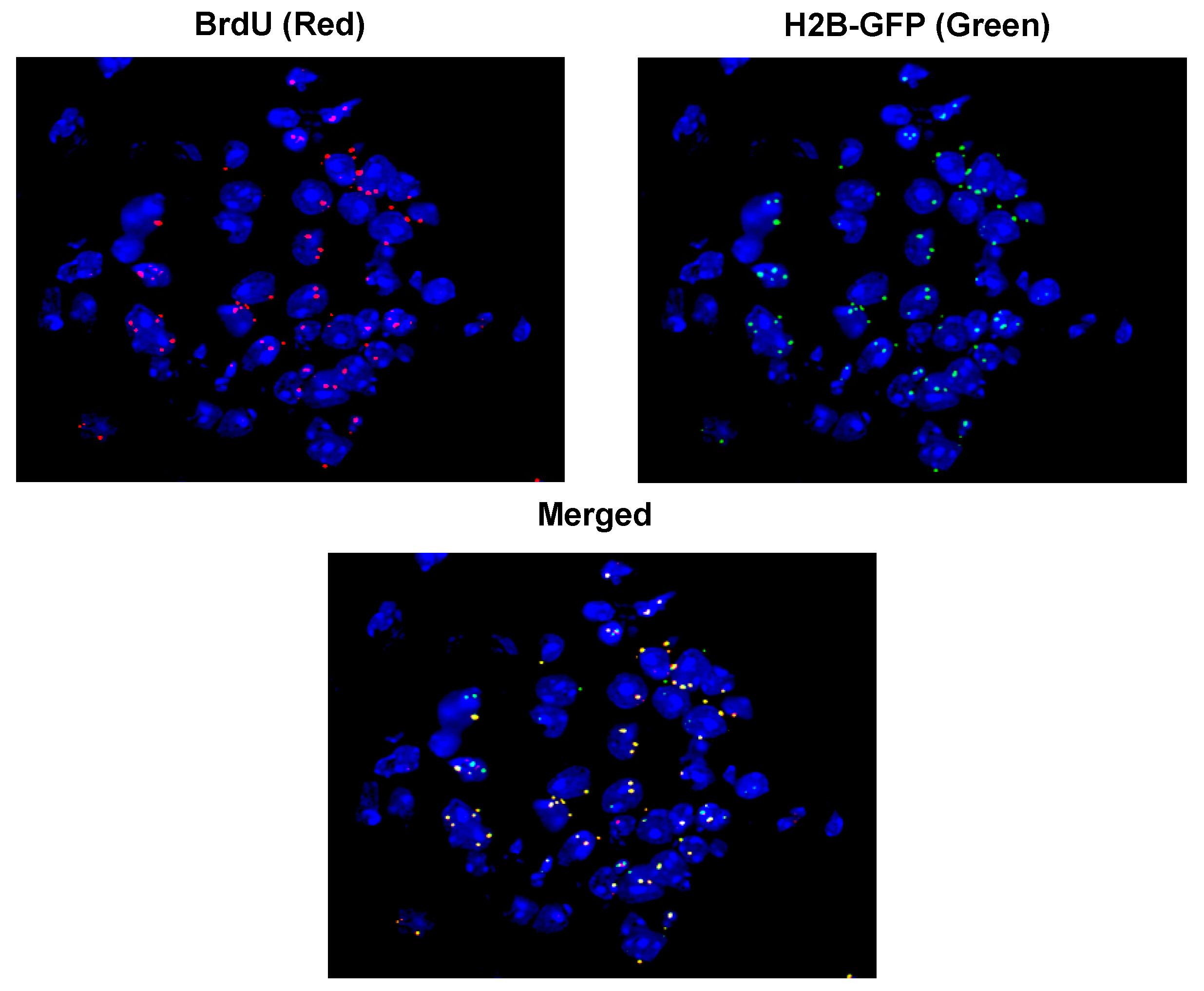
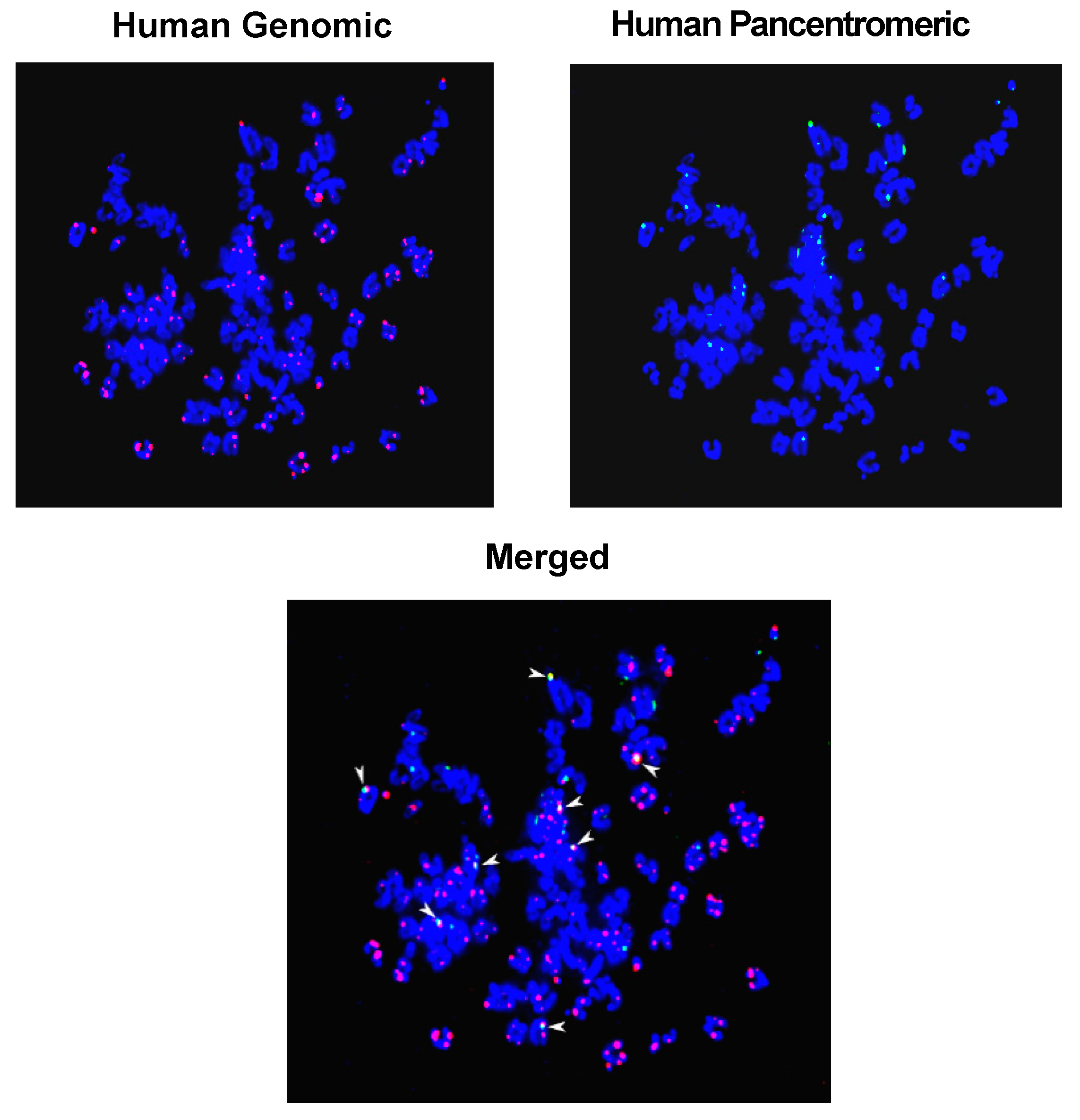
2.5. Mechanism of Genomic Integration of cfCh
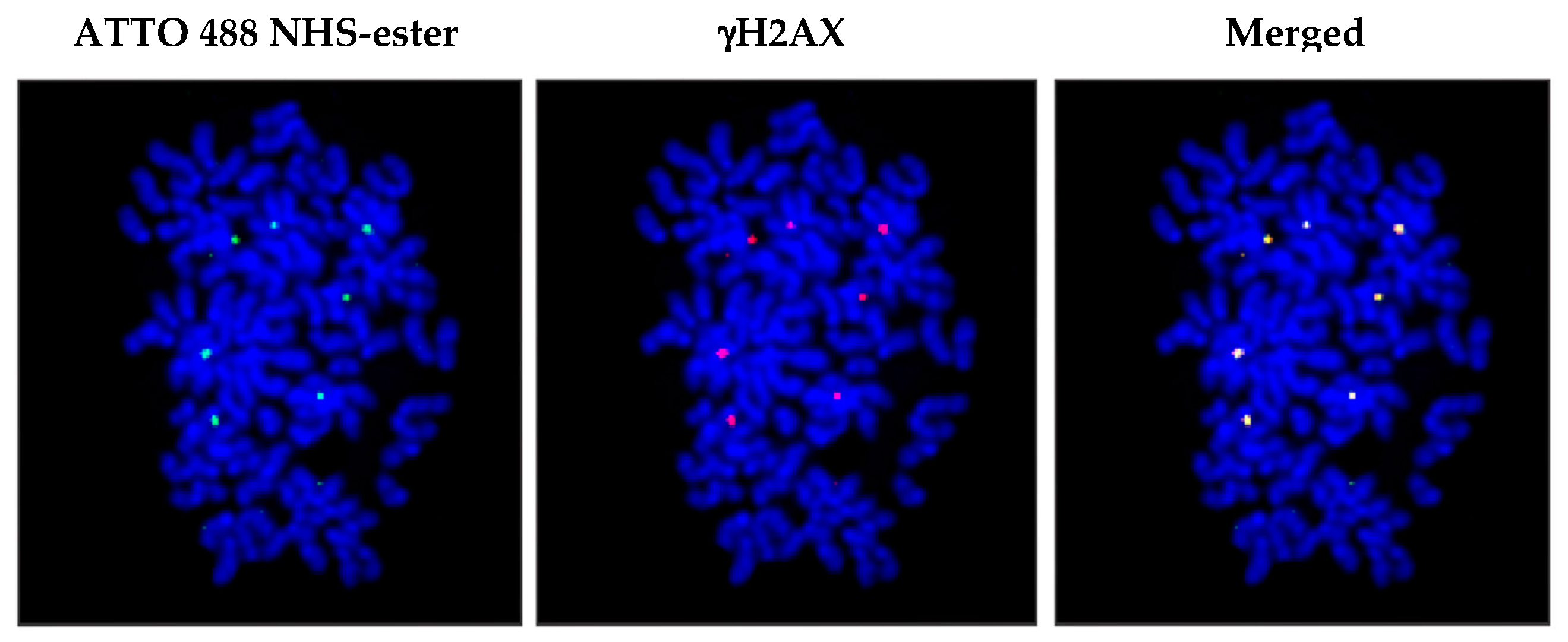
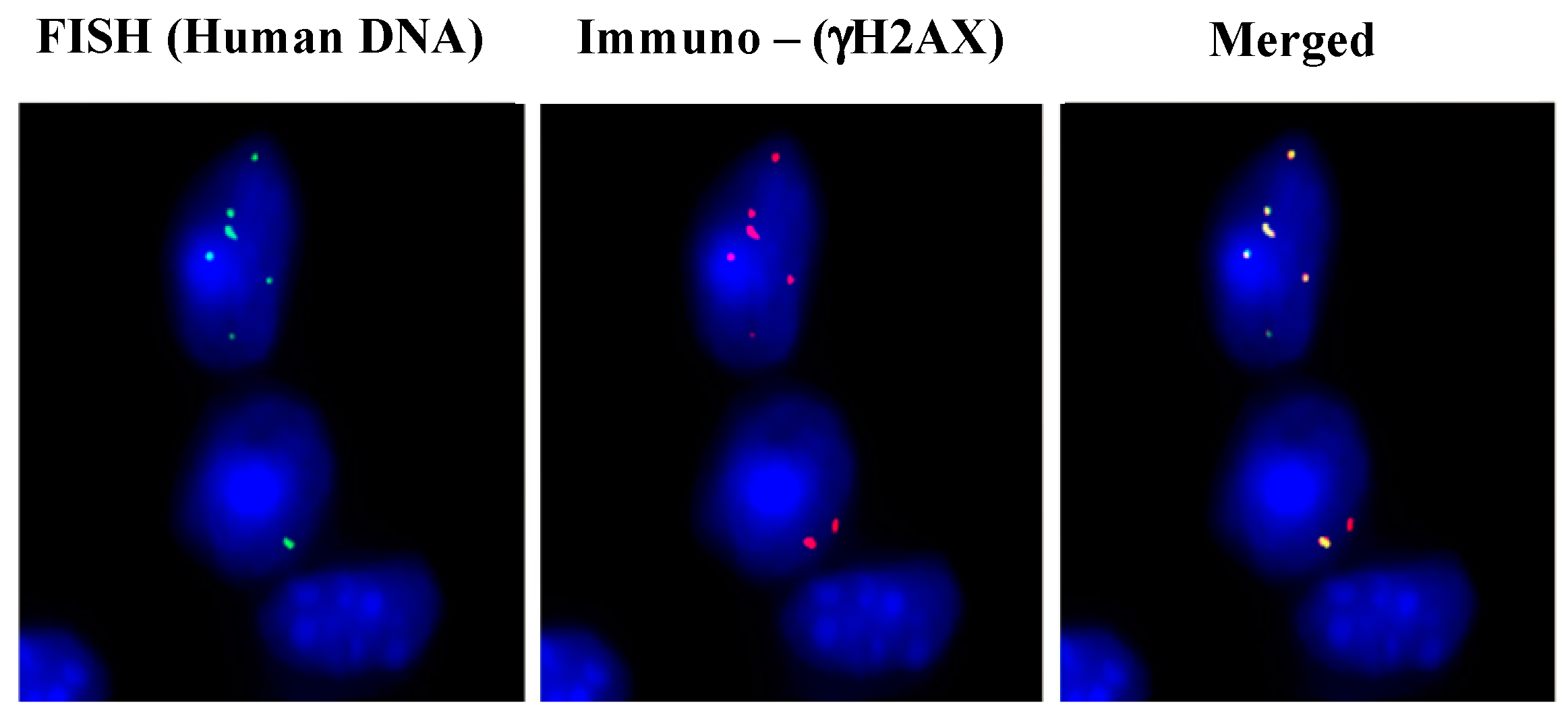
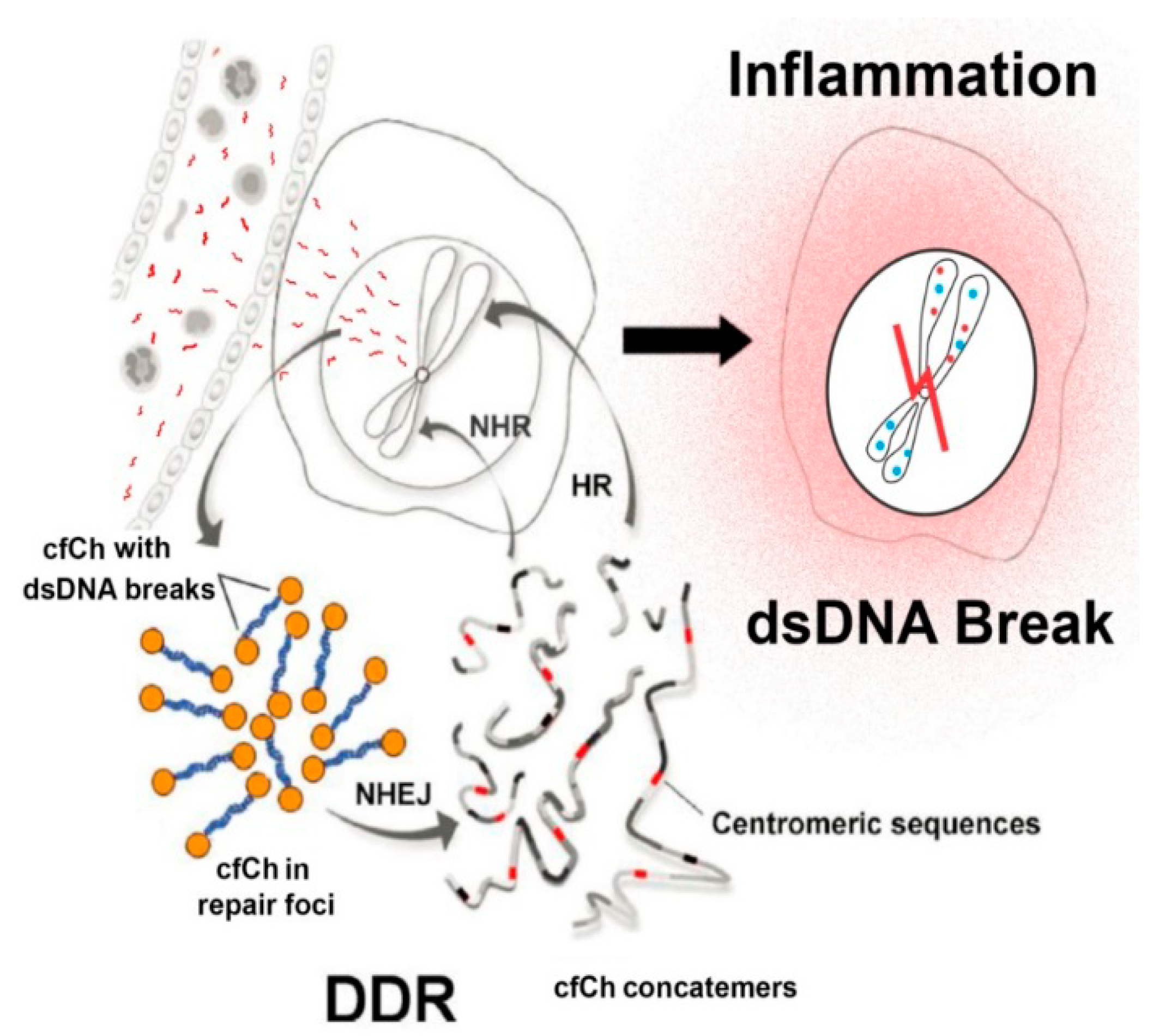
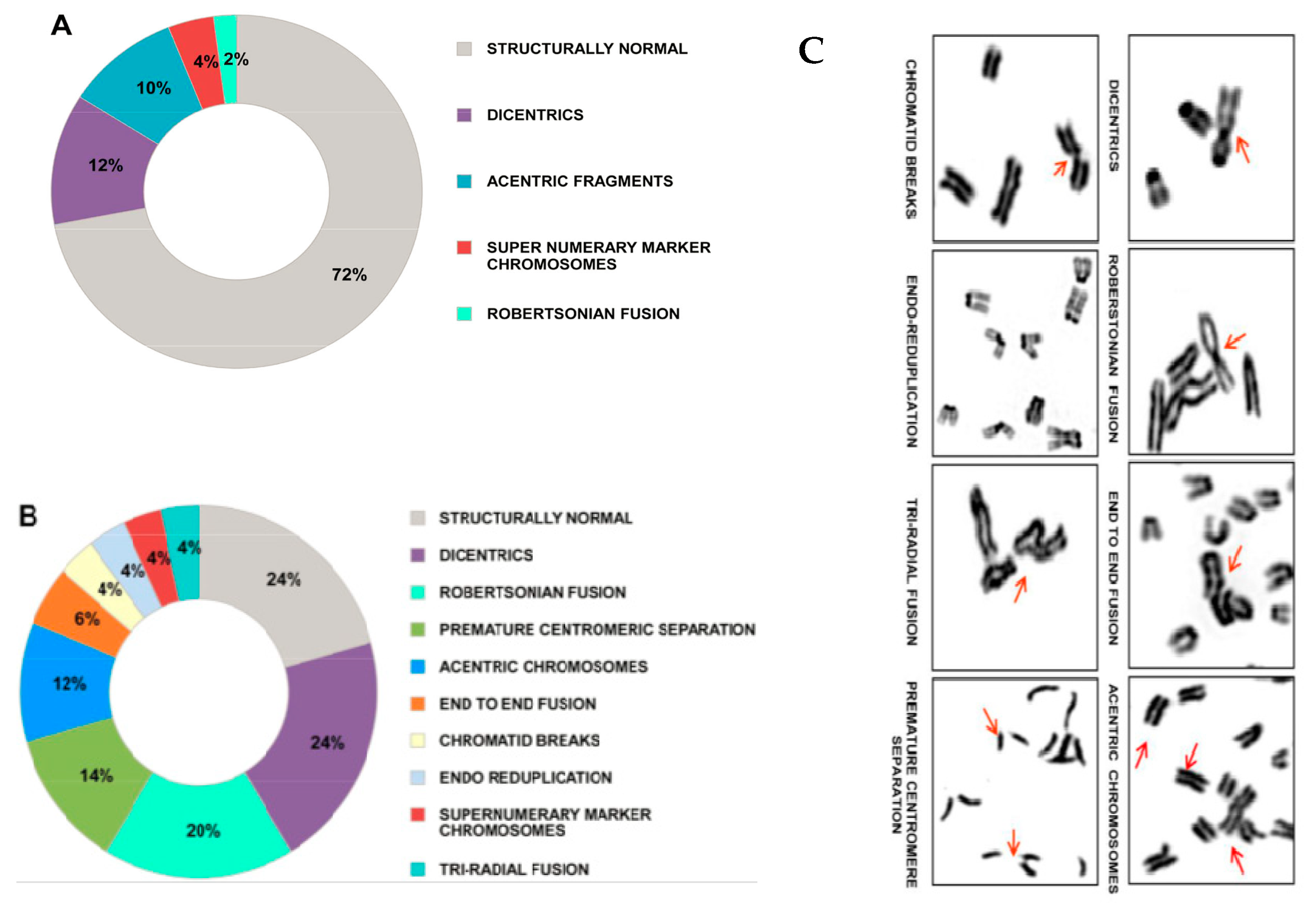
3. cfCh Integration, dsDNA Breaks and Somatic Mosaicism
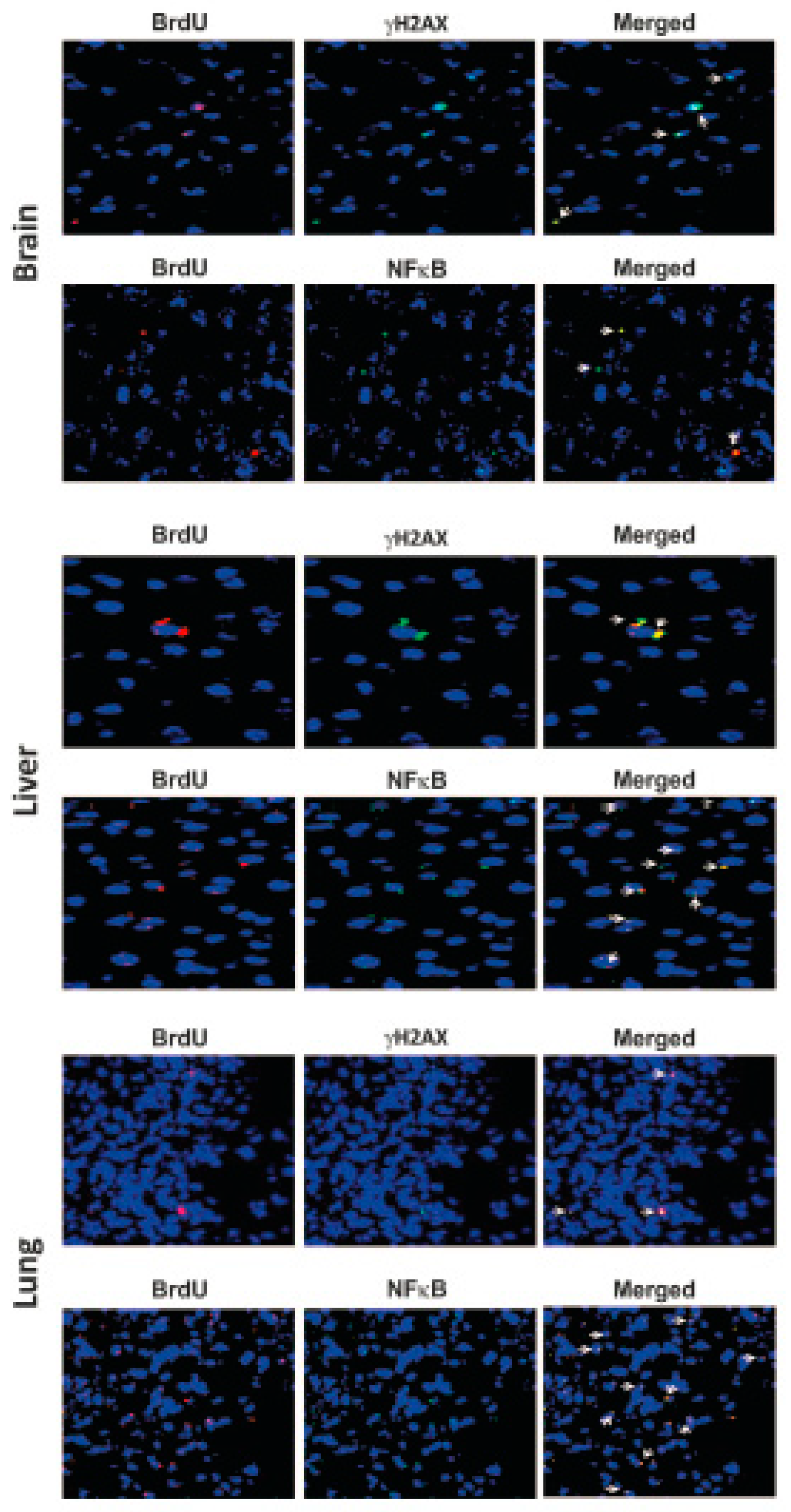
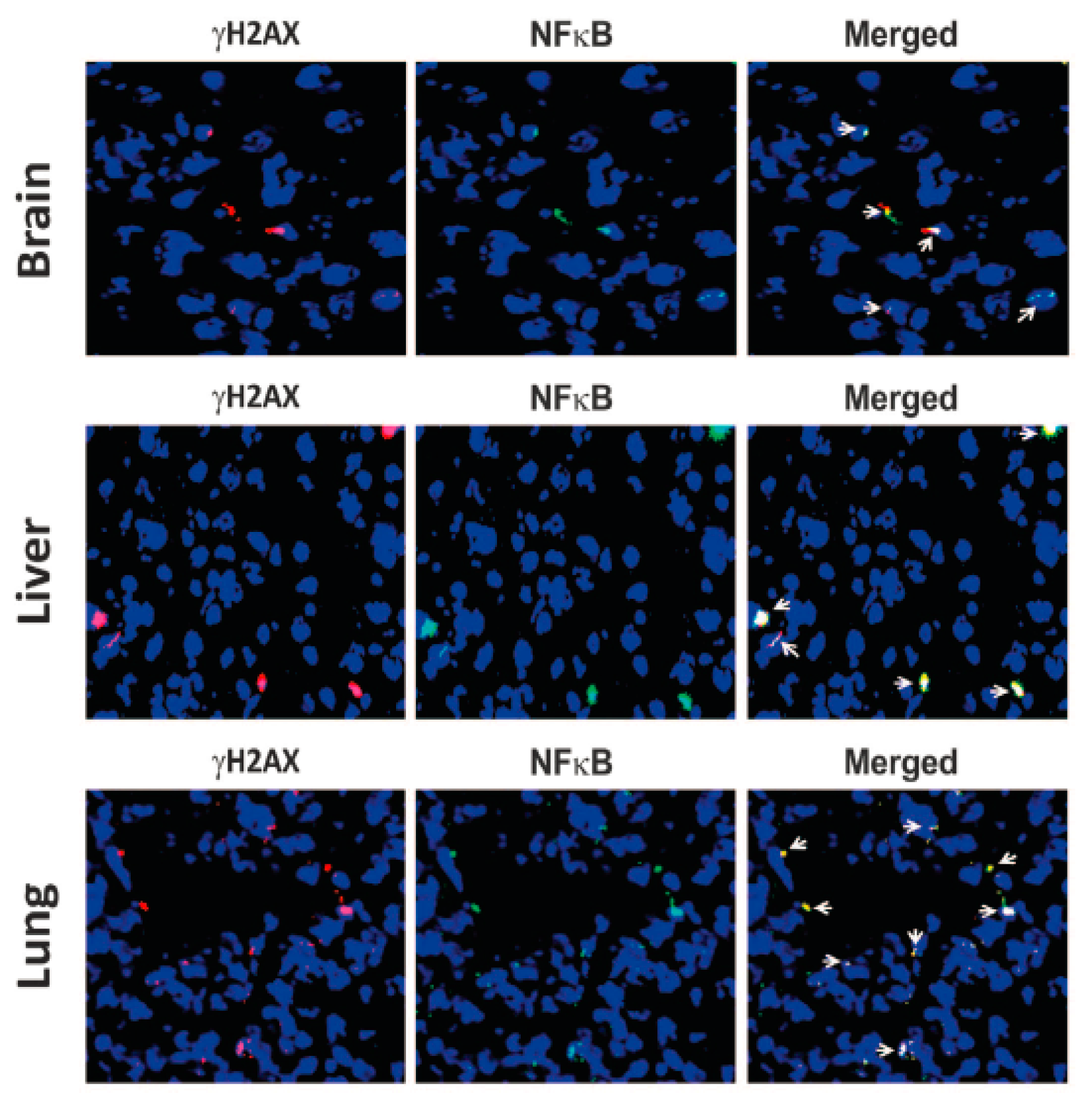
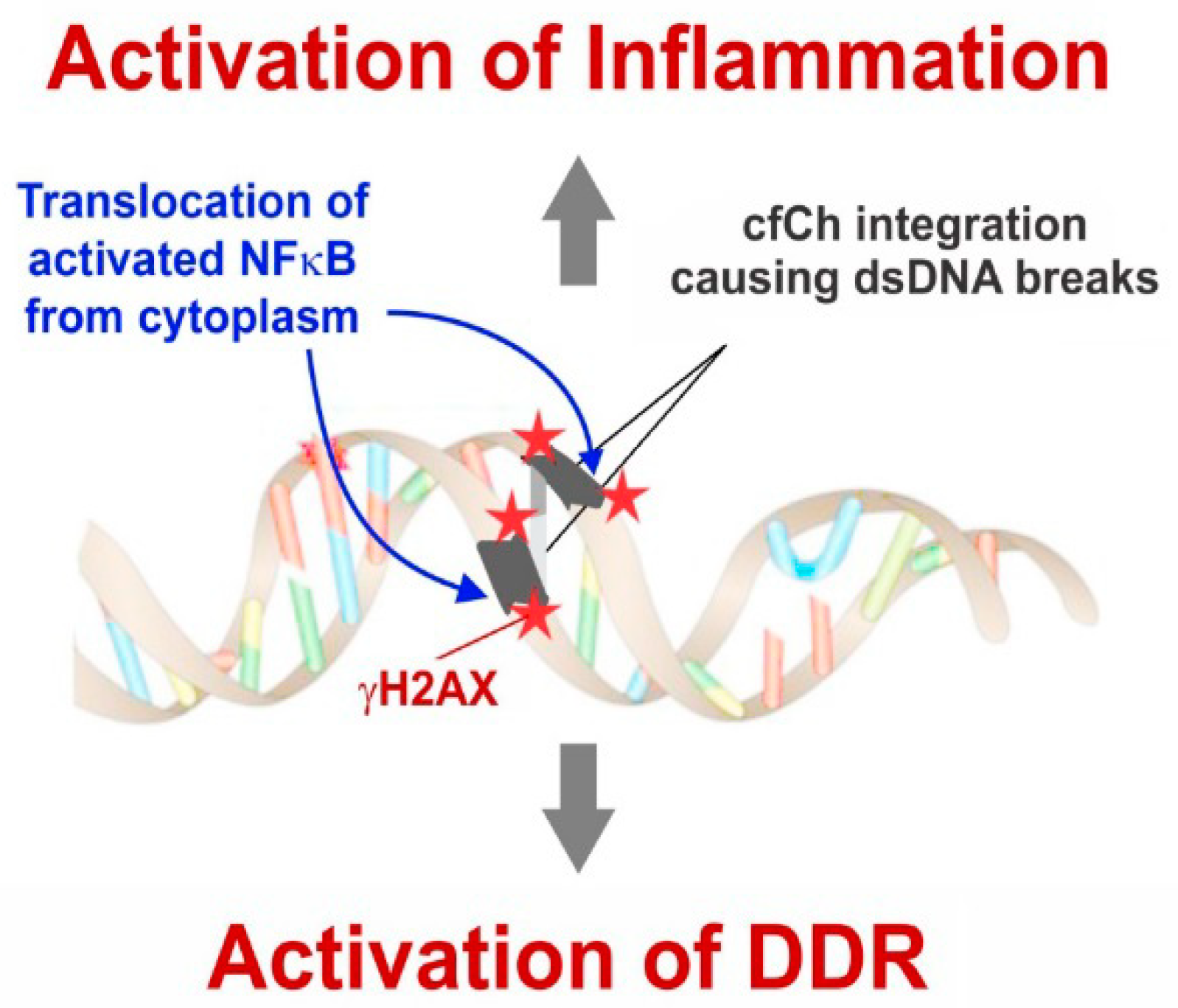
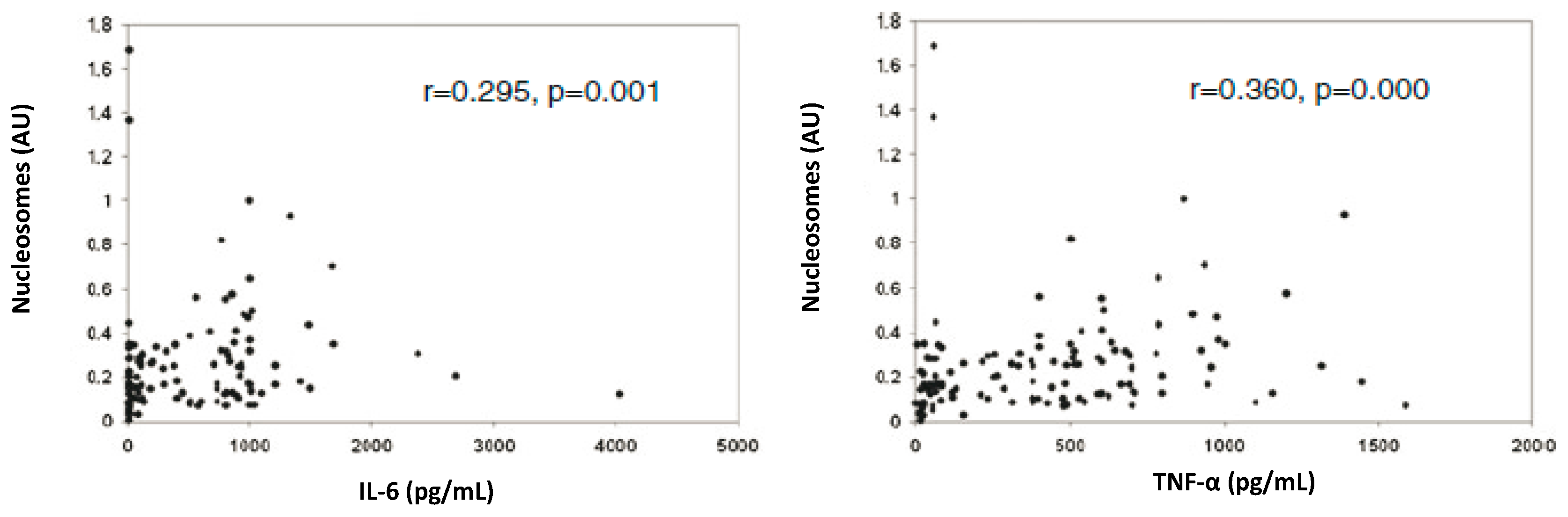
4. cfCh Integration, dsDNA Breaks and Inflammation
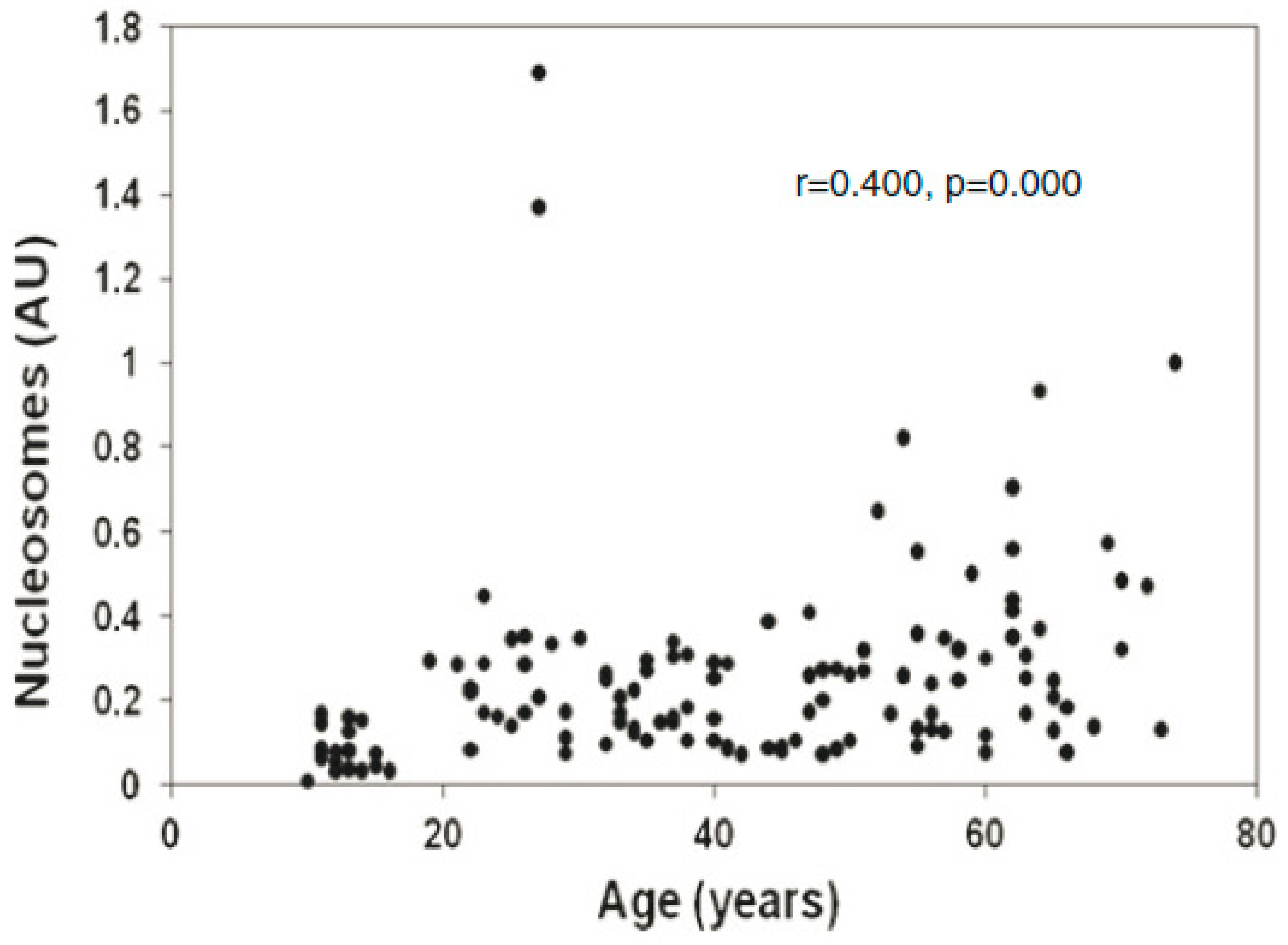
5. cfCh Induced Somatic Mosaicism, DNA Damage and Inflammation in Aetiology of Ageing, Chronic Diseases and Cancer
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Forsberg, L.A.; Gisselsson, D.; Dumanski, J.P. Mosaicism in health and disease-clones picking up speed. Nat. Rev. Genet. 2017, 18, 128–142. [Google Scholar] [CrossRef]
- Dou, Y.; Gold, H.D.; Luquette, L.J.; Park, P.J. Detecting somatic mutations in normal cells. Trends Genet. 2018, 34, 545–557. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, K.B.; Yeager, M.; Zhou, W.; Wacholder, S.; Wang, Z.; Rodriguez-Santiago, B.; Hutchinson, A.; Deng, X.; Liu, C.; Horner, M.J.; et al. Detectable clonal mosaicism and its relationship to aging and cancer. Nat. Genet. 2012, 44, 651–658. [Google Scholar] [CrossRef]
- McConnell, M.J.; Lindberg, M.R.; Brennand, K.J.; Piper, J.C.; Voet, T.; Cowing-Zitron, C.; Shumilina, S.; Lasken, R.S.; Vermeesch, J.R.; Hall, I.M.; et al. Mosaic copy number variation in human neurons. Science 2013, 342, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Iourov, I.Y.; Vorsanova, S.G.; Yurov, Y.B. Chromosomal mosaicism goes global. Mol. Cytogenet. 2008, 1, 26. [Google Scholar] [CrossRef]
- Villela, D.; Suemoto, C.K.; Leite, R.; Pasqualucci, C.A.; Grinberg, L.T.; Pearson, P.; Rosenberg, C. Increased DNA copy number variation mosaicism in elderly human brain. Neural Plast. 2018, 2018, 2406170. [Google Scholar] [CrossRef] [PubMed]
- Conlin, L.K.; Thiel, B.D.; Bonnemann, C.G.; Medne, L.; Ernst, L.M.; Zackai, E.H.; Deardorff, M.A.; Krantz, I.D.; Hakonarson, H.; Spinner, N.B. Mechanisms of mosaicism, chimerism and uniparental disomy identified by single nucleotide polymorphism array analysis. Hum. Mol. Genet. 2010, 19, 1263–1275. [Google Scholar] [CrossRef] [Green Version]
- Gajecka, M. Unrevealed mosaicism in the next-generation sequencing era. Mol. Genet. Genom. 2016, 291, 513–530. [Google Scholar] [CrossRef]
- King, D.A.; Sifrim, A.; Fitzgerald, T.W.; Rahbari, R.; Hobson, E.; Homfray, T.; Mansour, S.; Mehta, S.G.; Shehla, M.; Tomkins, S.E.; et al. Detection of structural mosaicism from targeted and whole-genome sequencing data. Genome Res. 2017, 10, 1704–1714. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Woo, C.J.; Iglesias-Ussel, M.D.; Ronai, D.; Scharff, M.D. The generation of antibody diversity through somatic hypermutation and class switch recombination. Genes Dev. 2004, 18, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Mkrtchyan, H.; Gross, M.; Hinreiner, S.; Polytiko, A.; Manvelyan, M.; Mrasek, K.; Kosyakova, N.; Ewers, E.; Nelle, H.; Liehr, T.; et al. The human genome puzzle—The role of copy number variation in somatic mosaicism. Curr. Genom. 2010, 11, 426–431. [Google Scholar] [CrossRef]
- Abyzov, A.; Tomasini, L.; Zhou, B.; Vasmatzis, N.; Coppola, G.; Amenduni, M.; Pattni, R.; Wilson, M.; Gerstein, M.; Weissman, S.; et al. One thousand somatic SNVs per skin fibroblast cell set baseline of mosaic mutational load with patterns that suggest proliferative origin. Genome Res. 2017, 27, 512–523. [Google Scholar] [CrossRef]
- Zhang, L.; Vijg, J. Somatic mutagenesis in mammals and its implications for human disease and aging. Annu. Rev. Genet. 2018, 52, 397–419. [Google Scholar] [CrossRef]
- Waris, G.; Ahsan, H. Reactive oxygen species: Role in the development of cancer and various chronic conditions. J. Carcinog. 2006, 5, 14. [Google Scholar] [CrossRef]
- Richardson, S.R.; Morell, S.; Faulkner, G.J. L1 retrotransposons and somatic mosaicism in the brain. Annu. Rev. Genet. 2014, 48, 1–27. [Google Scholar] [CrossRef]
- Upton, K.R.; Gerhardt, D.J.; Jesuadian, J.S.; Richardson, S.R.; Sánchez-Luque, F.J.; Bodea, G.O.; Ewing, A.D.; Salvador-Palomeque, C.; Van Der Knaap, M.S.; Brennan, P.M.; et al. Ubiquitous L1 mosaicism in hippocampal neurons. Cell 2015, 161, 228–239. [Google Scholar] [CrossRef]
- Kurnosov, A.A.; Ustyugova, S.V.; Nazarov, V.I.; Minervina, A.A.; Komkov, A.Y.; Shugay, M.; Pogorelyy, M.V.; Khodosevich, K.V.; Mamedov, I.Z.; Lebedev, Y.B. The evidence for increased L1 activity in the site of human adult brain neurogenesis. PLoS ONE 2015, 10, e01178540. [Google Scholar] [CrossRef]
- Usdin, K. The biological effects of simple tandem repeats: Lessons from the repeat expansion diseases. Genome Res. 2008, 18, 1011–1019. [Google Scholar] [CrossRef]
- Schmucker, B.; Ballhausen, W.G.; Pfeiffer, R.A. Mosaicism of a microdeletion of 486 bp involving the CGG repeat of the FMR1 gene due to misalignment of GTT tandem repeats at chi-like elements flanking both breakpoints and a full mutation. Hum. Genet. 1996, 98, 409–414. [Google Scholar] [CrossRef]
- Ghezraoui, H.; Piganeau, M.; Renouf, B.; Renaud, J.B.; Sallmyr, A.; Ruis, B.; Oh, S.; Tomkinson, A.E.; Hendrickson, E.A.; Giovannangeli, C.; et al. Chromosomal translocations in human cells are generated by canonical nonhomologous end-joining. Mol. Cell 2014, 55, 829–842. [Google Scholar] [CrossRef]
- Lee, J.A.; Carvalho, C.M.B.; Lupski, J.R. A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell 2007, 131, 1235–1247. [Google Scholar] [CrossRef] [PubMed]
- Giam, M.; Rancati, G. Aneuploidy and chromosomal instability in cancer: A jackpot to chaos. Cell Div. 2015, 10, 3. [Google Scholar] [CrossRef] [PubMed]
- Russel, L.M.; Strike, P.; Browne, C.E.; Jacobs, P.A. X chromosome loss and aging. Cytogenet. Genome Res. 2007, 116, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Geigl, J.B.; Langer, S.; Barwisch, S.; Pfleghaar, K.; Lederer, G.; Speicher, M.R. Analysis of gene expression patterns and chromosomal changes associated with aging. Cancer Res. 2004, 64, 8550–8557. [Google Scholar] [CrossRef] [PubMed]
- Invernizzi, P.; Miozzo, M.; Selmi, C.; Persani, L.; Battezzati, P.M.; Zuin, M.; Lucchi, S.; Meroni, P.L.; Marasini, B.; Zeni, S.; et al. X chromosome monosomy: A common mechanism for autoimmune diseases. J. Immunol. 2005, 175, 575–578. [Google Scholar] [CrossRef] [PubMed]
- Yurov, Y.B.; Vorsanova, S.G.; Iourov, I.Y.; Demidova, I.A.; Beresheva, A.K.; Kravetz, V.S.; Monakhov, V.V.; Kolotii, A.D.; Voinova-Ulas, V.Y.; Gorbachevskaya, N.L. Unexplained autism is frequently associated with low-level mosaic aneuploidy. J. Med. Genet. 2007, 44, 521–535. [Google Scholar] [CrossRef]
- Yurov, Y.B.; Iourov, I.Y.; Vorsanova, S.G.; Demidova, I.A.; Kravetz, V.S.; Beresheva, A.K.; Kolotii, A.D.; Monakchov, V.V.; Uranova, N.A.; Vostrikov, V.M.; et al. The schizophrenia brain exhibits low-level aneuploidy involving chromosome 1. Schizophr. Res. 2008, 98, 137–147. [Google Scholar] [CrossRef]
- Hook, E.B. Chromosome abnormalities and spontaneous fetal death following amniocentesis: Further data and associations with maternal age. Am. J. Hum. Genet. 1983, 35, 110–116. [Google Scholar] [CrossRef]
- Vorsanova, S.G.; Kolotii, A.D.; Iourov, I.Y.; Monakhov, V.V.; Kirillova, E.A.; Soloviev, I.V.; Yurov, Y.B. Evidence for high frequency of chromosomal mosaicism in spontaneous abortions revealed by interphase FISH analysis. J. Histochem. Cytochem. 2005, 53, 375–380. [Google Scholar] [CrossRef]
- Mosch, B.; Morawski, M.; Mittag, A.; Lenz, D.; Tarnok, A.; Arendt, T. Aneuploidy and DNA replication in the normal human brain and Alzheimer’s disease. J. Neurosci. 2007, 27, 6859–6867. [Google Scholar] [CrossRef]
- Ye, C.J.; Liu, G.; Bremer, S.W.; Heng, H.H.Q. The dynamics of cancer chromosomes and genomes. Cytogenet. Genome Res. 2007, 118, 237–246. [Google Scholar] [CrossRef]
- Forsberg, L.A. Loss of chromosome Y (LOY) in blood cells is associated with increased risk for disease and mortality in aging men. Hum. Genet. 2017, 136, 657–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forsberg, L.A.; Rasi, C.; Malmqvist, N.; Davies, H.; Pasupulati, S.; Pakalapati, G.; Sandgren, J.; De Ståhl, T.D.; Zaghlool, A.; Giedraitis, V.; et al. Mosaic loss of chromosome y in peripheral blood is associated with shorter survival and higher risk of cancer. Nat. Genet. 2014, 46, 624–628. [Google Scholar] [CrossRef] [PubMed]
- United Kingdom Cancer Cytogenetics Group (UKCCG). Loss of the Y chromosome from normal and neoplastic bone marrows. Genes Chromosom. Cancer 1992, 5, 83–88. [Google Scholar] [CrossRef]
- McConnell, M.J.; Moran, J.V.; Abyzov, A.; Akbarian, S.; Bae, T.; Cortes-Ciriano, I.; Erwin, J.A.; Fasching, L.; Flasch, D.A.; Freed, D.; et al. Intersection of diverse neuronal genomes and neuropsychiatric disease: The brain somatic mosaicism network. Science 2017, 356, eaal1641. [Google Scholar] [CrossRef] [PubMed]
- Rohrback, S.; April, C.; Kaper, F.; Rivera, R.R.; Liu, C.S.; Siddoway, B.; Chun, J. Submegabase copy number variations arise during cerebral cortical neurogenesis as revealed by single-cell whole-genome sequencing. Proc. Natl. Acad. Sci. USA 2018, 115, 10804–10809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, X.; Evrony, G.D.; Lehmann, H.S.; Elhosary, P.C.; Mehta, B.K.; Poduri, A.; Walsh, C.A. Single-cell, genome-wide sequencing identifies clonal somatic copy-number variation in the human brain. Cell Rep. 2014, 8, 1280–1289. [Google Scholar] [CrossRef]
- Lodato, M.A.; Woodworth, M.B.; Lee, S.; Evrony, G.D.; Mehta, B.K.; Karger, A.; Lee, S.; Chittenden, T.W.; D’Gama, A.M.; Cai, X.; et al. Somatic mutation in single human neurons tracks developmental and transcriptional history. Science 2015, 350, 94–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evrony, G.D.; Lee, E.; Park, P.J.; Walsh, C.A. Resolving rates of mutation in the brain using single-neuron genomics. Elife 2016, 5, e12966. [Google Scholar] [CrossRef]
- Lee, M.-H.; Siddoway, B.; Kaeser, G.E.; Segota, I.; Rivera, R.; Romanow, W.J.; Liu, C.S.; Park, C.; Kennedy, G.; Long, T.; et al. Somatic APP gene recombination in Alzheimer’s disease and normal neurons. Nature 2018, 563, 639–645. [Google Scholar] [CrossRef]
- Genovese, G.; Kähler, A.K.; Handsaker, R.E.; Lindberg, J.; Rose, S.A.; Bakhoum, S.F.; Chambert, K.; Mick, E.; Neale, B.M.; Fromer, M.; et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med. 2014, 371, 2477–2487. [Google Scholar] [CrossRef]
- Forsberg, L.A.; Rasi, C.; Razzaghian, H.R.; Pakalapati, G.; Waite, L.; Thilbeault, K.S.; Ronowicz, A.; Wineinger, N.E.; Tiwari, H.K.; Boomsma, D.; et al. Age-related somatic structural changes in the nuclear genome of human blood cells. Am. J. Hum. Genet. 2012, 90, 217–228. [Google Scholar] [CrossRef]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef]
- Laurie, C.C.; Laurie, C.A.; Rice, K.; Doheny, K.F.; Zelnick, L.R.; McHugh, C.P.; Ling, H.; Hetrick, K.N.; Pugh, E.W.; Amos, C.; et al. Detectable clonal mosaicism from birth to old age and its relationship to cancer. Nat. Genet. 2012, 44, 642–650. [Google Scholar] [CrossRef] [Green Version]
- Cooper, C.S.; Eeles, R.; Wedge, D.C.; Van Loo, P.; Gundem, G.; Alexandrov, L.B.; Kremeyer, B.; Butler, A.; Lynch, A.G.; Camacho, N.; et al. Analysis of the genetic phylogeny of multifocal prostate cancer identifies multiple independent clonal expansions in neoplastic and morphologically normal prostate tissue. Nat. Genet. 2015, 47, 367–372. [Google Scholar] [CrossRef]
- Yadav, V.K.; Degregori, J.; De, S. The landscape of somatic mutations in protein coding genes in apparently benign human tissues carries signatures of relaxed purifying selection. Nucleic Acids Res. 2016, 44, 2075–2084. [Google Scholar] [CrossRef]
- Bonnefond, A.; Skrobek, B.; Lobbens, S.; Eury, E.; Thuillier, D.; Cauchi, S.; Lantieri, O.; Balkau, B.; Riboli, E.; Marre, M.; et al. Association between large detectable clonal mosaicism and type 2 diabetes with vascular complications. Nat. Genet. 2013, 45, 1040–1043. [Google Scholar] [CrossRef]
- Dumanski, J.P.; Lambert, J.C.; Rasi, C.; Giedraitis, V.; Davies, H.; Grenier-Boley, B.; Lindgren, C.M.; Campion, D.; Dufouil, C.; Pasquier, F.; et al. Mosaic loss of chromosome Y in blood is associated with Alzheimer disease. Am. J. Hum. Genet. 2016, 98, 1208–1219. [Google Scholar] [CrossRef]
- Jacobs, K.; Mertzanidou, A.; Geens, M.; Thi Nguyen, H.; Staessen, C.; Spits, C. Low-grade chromosomal mosaicism in human somatic and embryonic stem cell populations. Nat. Commun. 2014, 5, 4227. [Google Scholar] [CrossRef] [Green Version]
- Martincorena, I.; Roshan, A.; Gerstung, M.; Ellis, P.; Van Loo, P.; McLaren, S.; Wedge, D.C.; Fullam, A.; Alexandrov, L.B.; Tubio, J.M.; et al. High burden and pervasive positive selection of somatic mutations in normal human skin. Science 2015, 348, 880–886. [Google Scholar] [CrossRef] [Green Version]
- Martincorena, I.; Fowler, J.C.; Wabik, A.; Lawson, A.R.; Abascal, F.; Hall, M.W.; Cagan, A.; Murai, K.; Mahbubani, K.; Stratton, M.R.; et al. Somatic mutant clones colonize the human esophagus with age. Science 2018, 362, 911–917. [Google Scholar] [CrossRef] [Green Version]
- Fliedner, T.M.; Graessle, D.; Paulsen, C.; Reimers, K. Structure and function of bone marrow hemopoiesis: Mechanisms of response to ionizing radiation exposure. Cancer Biother. Radiopharm. 2002, 17, 405–426. [Google Scholar] [CrossRef]
- Maslinska, D. Apoptosis: Physiological cell death and its role in pathogenesis of diseases. Neurol. Neurochir. Pol. 2003, 37, 315–326. [Google Scholar]
- Kerr, J.F.R.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 1972, 126, 608–614. [Google Scholar]
- Sakahira, H.; Enari, M.; Nagata, S. Cleavage of CAD inhibitor in CAD activation and DNA degradation during apoptosis. Nature 1998, 391, 96–99. [Google Scholar] [CrossRef]
- Wyllie, A.H. Glucocorticoid-induced thymocyte apoptosis is associated with endogenous endonuclease activation. Nature 1980, 284, 555–556. [Google Scholar] [CrossRef]
- Van Nieuwenhuijze, A.E.M.; Van Lopik, T.; Smeenk, R.J.T.; Aarden, L.A. Time between onset of apoptosis and release of nucleosomes from apoptotic cells: Putative implications for systemic lupus erythematosus. Ann. Rheum. Dis. 2003, 62, 10–14. [Google Scholar] [CrossRef]
- Van Der Vaart, M.; Pretorius, P.J. The origin of circulating free DNA. Clin. Chem. 2007, 53, 2215. [Google Scholar] [CrossRef]
- Suzuki, N.; Kamataki, A.; Yamaki, J.; Homma, Y. Characterization of circulating DNA in healthy human plasma. Clin. Chim. Acta 2008, 387, 55–58. [Google Scholar] [CrossRef]
- Holdenrieder, S.; Nagel, D.; Schalhorn, A.; Heinemann, V.; Wilkowski, R.; von Pawel, J.; Raith, H.; Feldmann, K.; Kremer, A.E.; Müller, S.; et al. Clinical relevance of circulating nucleosomes in cancer. Ann. N. Y. Acad. Sci. 2008, 1137, 180–189. [Google Scholar] [CrossRef]
- Jylhävä, J.; Kotipelto, T.; Raitala, A.; Jylhä, M.; Hervonen, A.; Hurme, M. Aging is associated with quantitative and qualitative changes in circulating cell-free DNA: The vitality 90+ study. Mech. Ageing Dev. 2011, 132, 20–26. [Google Scholar] [CrossRef]
- Mittra, I.; Nair, N.K.; Mishra, P.K. Nucleic acids in circulation: Are they harmful to the host? J. Biosci. 2012, 37, 301–312. [Google Scholar] [CrossRef]
- Giacona, M.B.; Ruben, G.C.; Iczkowski, K.A.; Roos, T.B.; Porter, D.M.; Sorenson, G.D. Cell-free DNA in human blood plasma: Length measurements in patients with pancreatic cancer and healthy controls. Pancreas 1998, 17, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Mittra, I.; Khare, N.K.; Raghuram, G.V.; Chaubal, R.; Khambatti, F.; Gupta, D.; Gaikwad, A.; Prasannan, P.; Singh, A.; Iyer, A.; et al. Circulating nucleic acids damage DNA of healthy cells by integrating into their genomes. J. Biosci. 2015, 40, 91–111. [Google Scholar] [CrossRef] [PubMed]
- Savill, J.; Gregory, C.; Haslett, C. Cell biology. Eat me or die. Science 2003, 302, 1516–1517. [Google Scholar] [CrossRef] [PubMed]
- Tamkovich, S.N.; Cherepanova, A.V.; Kolesnikova, E.V.; Rykova, E.Y.; Pyshnyi, D.V.; Vlassov, V.V.; Laktionov, P.P. Circulating DNA and DNase activity in human blood. Ann. N. Y. Acad. Sci. 2006, 1075, 191–196. [Google Scholar] [CrossRef]
- Cherepanova, A.V.; Tamkovich, S.N.; Bryzgunova, O.E.; Vlassov, V.V.; Laktionov, P.P. Deoxyribonuclease activity and circulating DNA concentration in blood plasma of patients with prostate tumors. Ann. N. Y. Acad. Sci. 2008, 1137, 218–221. [Google Scholar] [CrossRef]
- Gauthier, V.J.; Tyler, L.N.; Mannik, M. Blood clearance kinetics and liver uptake of mononucleosomes in mice. J. Immunol. 1996, 156, 1151–1156. [Google Scholar] [PubMed]
- Du Clos, T.W.; Volzer, M.A.; Hahn, F.F.; Xiao, R.; Mold, C.; Searles, R.P. Chromatin clearance in C57B1/10 mice: Interaction with heparan sulphate proteoglycans and receptors on Kupffer cells. Clin. Exp. Immunol. 1999, 117, 403–411. [Google Scholar] [CrossRef]
- Babayan, A.; Pantel, K. Advances in liquid biopsy approaches for early detection and monitoring of cancer. Genome Med. 2018, 10, 21. [Google Scholar] [CrossRef] [PubMed]
- Dewey, W.C.; Ling, C.C.; Meyn, R.E. Radiation-induced apoptosis: Relevance to radiotherapy. Int. J. Radiat. Oncol. Biol. Phys. 1995, 33, 781–796. [Google Scholar] [CrossRef]
- Holdenrieder, S.; Stieber, P.; Chan, L.Y.S.; Geiger, S.; Kremer, A.; Nagel, D.; Lo, Y.M.D. Cell-free DNA in serum and plasma: Comparison of ELISA and quantitative PCR. Clin. Chem. 2005, 51, 1544–1546. [Google Scholar] [CrossRef]
- Holdenrieder, S.; Stieber, P.; Bodenmüller, H.; Fertig, G.; Fürst, H.; Schmeller, N.; Untch, M.; Seidel, D. Nucleosomes in serum as a marker for cell death. Clin. Chem. Lab. Med. 2001, 39, 596–605. [Google Scholar] [CrossRef]
- Bhargava, P.M.; Shanmugam, G. Uptake of nonviral nucleic acids by mammalian cells. Prog. Nucleic Acid Res. Mol. Biol. 1971, 11, 103–192. [Google Scholar]
- Thierry, A.R.; El Messaoudi, S.; Gahan, P.B.; Anker, P.; Stroun, M. Origins, structures, and functions of circulating DNA in oncology. Cancer Metastasis Rev. 2016, 35, 347–376. [Google Scholar] [CrossRef] [Green Version]
- Wagstaff, K.M.; Fan, J.Y.; De Jesus, M.A.; Tremethick, D.J.; Jans, D.A. Efficient gene delivery using reconstituted chromatin enhanced for nuclear targeting. FASEB J. 2008, 22, 2232–2242. [Google Scholar] [CrossRef]
- Rumore, P.M.; Steinman, C.R. Endogenous circulating DNA in systemic lupus erythematosus. Occurrence as multimeric complexes bound to histone. J. Clin. Investig. 1990, 86, 69–74. [Google Scholar] [CrossRef]
- Mittra, I. Circulating nucleic acids: A new class of physiological mobile genetic elements. F1000 Res. 2015, 4, 924. [Google Scholar] [CrossRef]
- Basak, R.; Nair, N.K.; Mittra, I. Evidence for cell-free nucleic acids as continuously arising endogenous DNA mutagens. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2016, 793, 15–21. [Google Scholar] [CrossRef]
- Rekha, M.R.; Pal, K.; Bala, P.; Shetty, M.; Mittra, I.; Bhuvaneshwar, G.S.; Sharma, C.P. Pullulan-histone antibody nanoconjugates for the removal of chromatin fragments from systemic circulation. Biomaterials 2013, 34, 6328–6338. [Google Scholar] [CrossRef]
- Erwig, L.P.; Henson, P.M. Clearance of apoptotic cells by phagocytes. Cell Death Differ. 2008, 15, 243–250. [Google Scholar] [CrossRef]
- Kawane, K.; Ohtani, M.; Miwa, K.; Kizawa, T.; Kanbara, Y.; Yoshioka, Y.; Yoshikawa, H.; Nagata, S. Chronic polyarthritis caused by mammalian DNA that escapes from degradation in macrophages. Nature 2006, 443, 998–1002. [Google Scholar] [CrossRef]
- Mittra, I.; Samant, U.; Sharma, S.; Raghuram, G.V.; Saha, T.; Tidke, P.; Pancholi, N.; Gupta, D.; Prasannan, P.; Gaikwad, A.; et al. Cell-free chromatin from dying cancer cells integrate into genomes of bystander healthy cells to induce DNA damage and inflammation. Cell Death Discov. 2017, 3, 17015. [Google Scholar] [CrossRef] [Green Version]
- Kirolikar, S.; Prasannan, P.; Raghuram, G.V.; Pancholi, N.; Saha, T.; Tidke, P.; Chaudhari, P.; Shaikh, A.; Rane, B.; Pandey, R.; et al. Prevention of radiation-induced bystander effects by agents that inactivate cell-free chromatin released from irradiated dying cells. Cell Death Dis. 2018, 9, 1142. [Google Scholar] [CrossRef] [Green Version]
- Subramaniam, S.; Vohra, I.; Iyer, A.; Nair, N.K.; Mittra, I. A paradoxical synergism between resveratrol and copper (II) with respect to degradation of DNA and RNA. F1000 Res. 2015, 4, 1145. [Google Scholar] [CrossRef]
- Fidler, I.J. Metastasis: Quantitative analysis of distribution and fate of tumor emboli labeled with 125l-5-lodo-2′-deoxyuridine. J. Natl. Cancer Inst. 1970, 45, 773–782. [Google Scholar]
- Kim, J.W.; Wong, C.W.; Goldsmith, J.D.; Song, C.; Fu, W.; Allion, M.B.; Herlyn, M.; Al-Mehdi, A.B.; Muschel, R.J. Rapid apoptosis in the pulmonary vasculature distinguishes non-metastatic from metastatic melanoma cells. Cancer Lett. 2004, 213, 203–212. [Google Scholar] [CrossRef]
- Frengen, E.; Thomsen, P.D.; Brede, G.; Solheim, J.; De Jong, P.J.; Prydz, H. The gene cluster containing the LCAT gene is conserved between human and pig. Cytogenet. Cell Genet. 1997, 76, 53–57. [Google Scholar] [CrossRef]
- Espejel, S.; Franco, S.; Rodríguez-Perales, S.; Bouffler, S.D.; Cigudosa, J.C.; Blasco, M.A. Mammalian Ku86 mediates chromosomal fusions and apoptosis caused by critically short telomeres. EMBO J. 2002, 21, 2207–2219. [Google Scholar] [CrossRef]
- Guirouilh-Barbat, J.; Huck, S.; Bertrand, P.; Pirzio, L.; Desmaze, C.; Sabatier, L.; Lopez, B.S. Impact of the KU80 pathway on NHEJ-induced genome rearrangements in mammalian cells. Mol. Cell 2004, 14, 611–623. [Google Scholar] [CrossRef]
- Heidenreich, E.; Novotny, R.; Kneidinger, B.; Holzmann, V.; Wintersberger, U. Non-homologous end joining as an important mutagenic process in cell cycle-arrested cells. EMBO J. 2003, 22, 2274–2283. [Google Scholar] [CrossRef] [Green Version]
- Yurov, Y.B.; Iourov, I.Y.; Vorsanova, S.G.; Liehr, T.; Kolotii, A.D.; Kutsev, S.I.; Pellestor, F.; Beresheva, A.K.; Demidova, I.A.; Kravets, V.S.; et al. Aneuploidy and confined chromosomal mosaicism in the developing human brain. PLoS ONE 2007, 2, e558. [Google Scholar] [CrossRef]
- Pahl, H. Activators and target genes of Rel/NF-κB transcription factors. Oncogene 1999, 18, 6853–6866. [Google Scholar] [CrossRef]
- Hadian, K.; Krappmann, D. Signals from the nucleus: Activation of NF-κB by cytosolic ATM in the DNA damage response. Sci. Signal. 2011, 4, pe2. [Google Scholar] [CrossRef]
- Wan, F.; Lenardo, M.J. Specification of DNA binding activity of NF-κB proteins. Cold Spring Harb. Perspect. Biol. 2009, a000067. [Google Scholar] [CrossRef]
- Chaudhary, S.; Raghuram, G.V.; Mittra, I. Is inflammation a direct response to dsDNA breaks? Mutat. Res. Fundam. Mol. Mech. Mutagen. 2018, 808, 48–52. [Google Scholar] [CrossRef]
- Härtlova, A.; Erttmann, S.F.; Raffi, F.A.M.; Schmalz, A.M.; Resch, U.; Anugula, S.; Lienenklaus, S.; Nilsson, L.M.; Kröger, A.; Nilsson, J.A.; et al. DNA damage primes the type I interferon system via the cytosolic DNA sensor STING to promote anti-microbial innate immunity. Immunity 2015, 42, 332–343. [Google Scholar] [CrossRef]
- Erdal, E.; Haider, S.; Rehwinkel, J.; Harris, A.L.; McHugh, P.J. A prosurvival DNA damage-induced cytoplasmic interferon response is mediated by end resection factors and is limited by Trex1. Genes Dev. 2017, 31, 353–369. [Google Scholar] [CrossRef] [Green Version]
- Dou, Z.; Ghosh, K.; Vizioli, M.G.; Zhu, J.; Sen, P.; Wangensteen, K.J.; Simithy, J.; Lan, Y.; Lin, Y.; Zhou, Z.; et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 2017, 550, 402–406. [Google Scholar] [CrossRef] [Green Version]
- Glück, S.; Guey, B.; Gulen, M.F.; Wolter, K.; Kang, T.W.; Schmacke, N.A.; Bridgeman, A.; Rehwinkel, J.; Zender, L.; Ablasser, A. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat. Cell Biol. 2017, 19, 1061–1107. [Google Scholar] [CrossRef]
- Yang, H.; Wang, H.; Ren, J.; Chen, Q.; Chen, Z.J. cGAS is essential for cellular senescence. Proc. Natl. Acad. Sci. USA 2017, 114, E4612–E4620. [Google Scholar] [CrossRef] [Green Version]
- Barber, G.N. STING: Infection, inflammation and cancer. Nat. Rev. Immunol. 2015, 15, 760–770. [Google Scholar] [CrossRef]
- Ishida, T.; Ishida, M.; Tashiro, S.; Yoshizumi, M.; Kihara, Y. Role of DNA damage in cardiovascular disease. Circ. J. 2014, 78, 42–50. [Google Scholar] [CrossRef]
- Ferrucci, L.; Fabbri, E. Inflammageing: Chronic inflammation in ageing, cardiovascular disease, and frailty. Nat. Rev. Cardiol. 2018, 15, 505–522. [Google Scholar] [CrossRef]
- Coppede, F.; Migliore, L. DNA damage and repair in Alzheimers disease. Curr. Alzheimer Res. 2009, 6, 36–47. [Google Scholar] [CrossRef]
- Wyss-Coray, T.; Rogers, J. Inflammation in Alzheimer disease—A brief review of the basic science and clinical literature. Cold Spring Harb. Perspect. Med. 2012, 2, a006346. [Google Scholar] [CrossRef]
- Blasiak, J.; Arabski, M.; Krupa, R.; Wozniak, K.; Zadrozny, M.; Kasznicki, J.; Zurawska, M.; Drzewoski, J. DNA damage and repair in type 2 diabetes mellitus. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2004, 554, 297–304. [Google Scholar] [CrossRef]
- Donath, M.Y.; Shoelson, S.E. Type 2 diabetes as an inflammatory disease. Nat. Rev. Immunol. 2011, 11, 98–107. [Google Scholar] [CrossRef]
- O’Connor, M.J. Targeting the DNA damage response in cancer. Mol. Cell 2015, 60, 547–560. [Google Scholar] [CrossRef]
- Mantovani, A.; Mantovani, A.; Allavena, P.; Allavena, P.; Sica, A.; Sica, A.; Balkwill, F.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Soares, J.P.; Silva, A.M.; Fonseca, S.; Oliveira, M.M.; Peixoto, F.; Gaivão, I.; Mota, M.P. How can age and lifestyle variables affect DNA damage, repair capacity and endogenous biomarkers of oxidative stress? Exp. Gerontol. 2015, 62, 45–52. [Google Scholar] [CrossRef]
- Franceschi, C.; Garagnani, P.; Vitale, G.; Capri, M.; Salvioli, S. Inflammaging and ‘Garb-aging’. Trends Endocrinol. Metab. 2017, 28, 199–212. [Google Scholar] [CrossRef]













© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
V. Raghuram, G.; Chaudhary, S.; Johari, S.; Mittra, I. Illegitimate and Repeated Genomic Integration of Cell-Free Chromatin in the Aetiology of Somatic Mosaicism, Ageing, Chronic Diseases and Cancer. Genes 2019, 10, 407. https://doi.org/10.3390/genes10060407
V. Raghuram G, Chaudhary S, Johari S, Mittra I. Illegitimate and Repeated Genomic Integration of Cell-Free Chromatin in the Aetiology of Somatic Mosaicism, Ageing, Chronic Diseases and Cancer. Genes. 2019; 10(6):407. https://doi.org/10.3390/genes10060407
Chicago/Turabian StyleV. Raghuram, Gorantla, Shahid Chaudhary, Shweta Johari, and Indraneel Mittra. 2019. "Illegitimate and Repeated Genomic Integration of Cell-Free Chromatin in the Aetiology of Somatic Mosaicism, Ageing, Chronic Diseases and Cancer" Genes 10, no. 6: 407. https://doi.org/10.3390/genes10060407
APA StyleV. Raghuram, G., Chaudhary, S., Johari, S., & Mittra, I. (2019). Illegitimate and Repeated Genomic Integration of Cell-Free Chromatin in the Aetiology of Somatic Mosaicism, Ageing, Chronic Diseases and Cancer. Genes, 10(6), 407. https://doi.org/10.3390/genes10060407