Temporal Analysis of the Microbial Community from the Crystallizer Ponds in Cabo Rojo, Puerto Rico, Using Metagenomics


Abstract
:1. Introduction
2. Materials and Methods
2.1. Sampling and DNA Purification
2.2. DNA Sequencing and Metagenome Assembly
2.3. Binning for Putative Genomes
3. Results and Discussion
3.1. Sampling Site Conditions and Sequencing Analysis
3.2. Microbial Community Composition
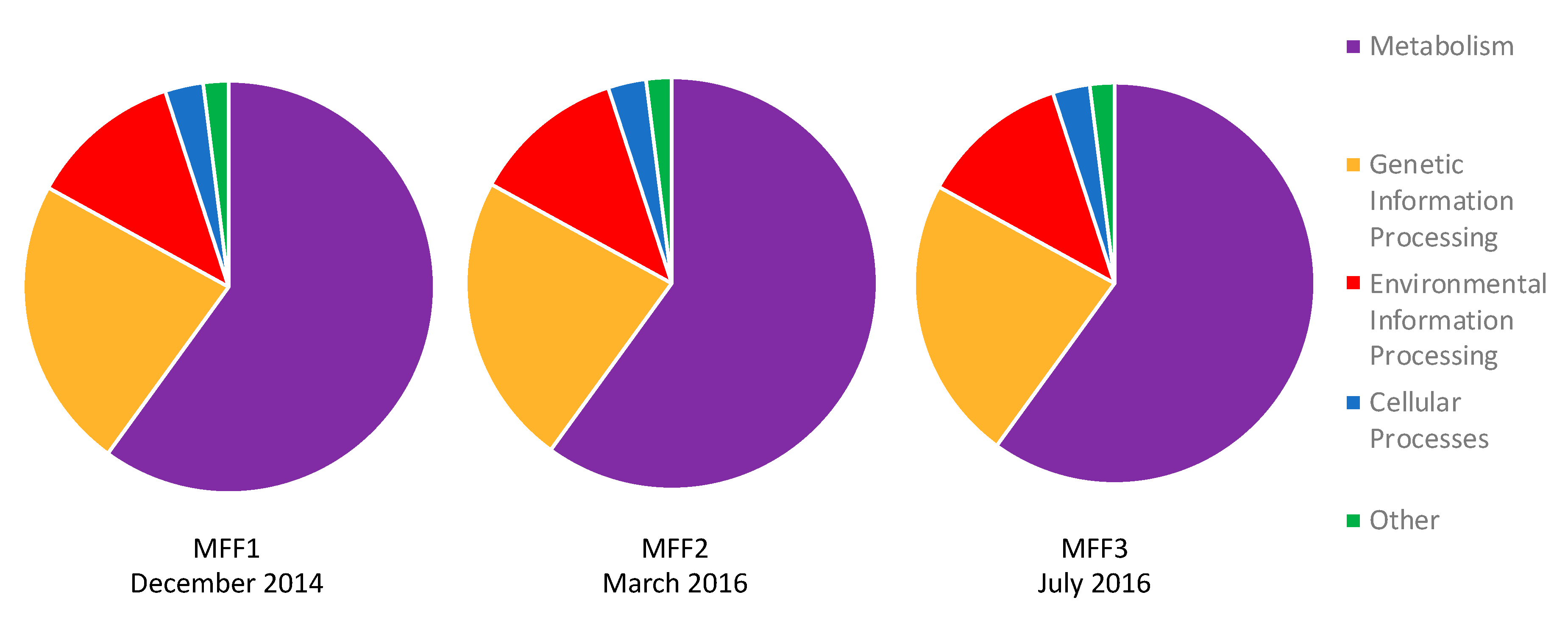
3.3. Functional Annotation
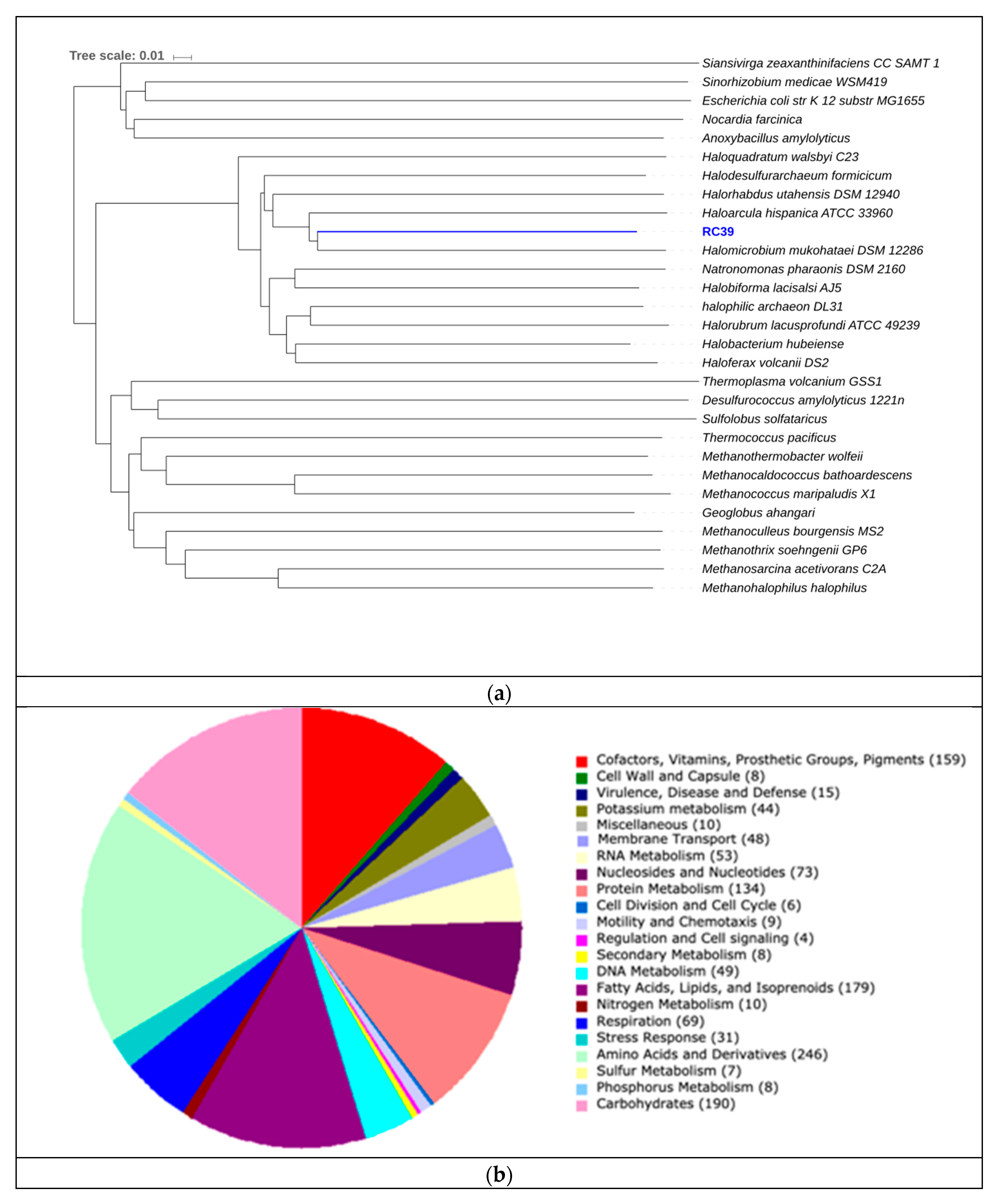
3.4. Binning of Putative Novel Genomes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Oren, A. Diversity of halophilic microorganisms: Environments, phylogeny, physiology, and applications. J. Ind. Microbiol. 2002, 28, 56–63. [Google Scholar]
- Ventosa, A. Unusual micro-organisms from unusual habitats: Hypersaline environments. In Symposia-Society for General Microbiology; Cambridge University Press: Cambridge, UK, 2006; Volume 66, p. 223. [Google Scholar]
- Sanchez-Porro, C.; Martı, S.; Mellado, E.; Ventosa, A. Diversity of moderately halophilic bacteria producing extracellular hydrolytic enzymes. J. Appl. Microbiol. 2003, 94, 295–300. [Google Scholar] [CrossRef] [PubMed]
- De Lourdes Moreno, M.; Pérez, D.; García, M.T.; Mellado, E. Halophilic bacteria as a source of novel hydrolytic enzymes. Life 2013, 3, 38–51. [Google Scholar] [CrossRef] [PubMed]
- Amoozegar, M.A.; Siroosi, M.; Atashgahi, S.; Smidt, H.; Ventosa, A. Systematics of Haloarchaea and Biotechnological Potential of their Hydrolytic Enzymes. Microbiology 2017, 163, 623–645. [Google Scholar] [CrossRef] [PubMed]
- Ozcan, B.; Ozcengiz, G.; Coleri, A.; Cokmus, C. Diversity of Halophilic Archaea From Six Hypersaline Environments in Turkey. J. Microbiol. Biotechnol. 2007, 17, 985–992. [Google Scholar] [PubMed]
- Ghai, R.; Pašić, L.; Fernández, A.B.; Martin-Cuadrado, A.-B.; Mizuno, C.M.; McMahon, K.D.; Papke, R.T.; Stepanauskas, R.; Rodriguez-Brito, B.; Rohwer, F.; et al. New abundant microbial groups in aquatic hypersaline environments. Sci. Rep. 2011, 1, 135. [Google Scholar] [CrossRef]
- Ghai, R.; Hernandez, C.M.; Picazo, A.; Mizuno, C.M.; Ininbergs, K.; Díez, B.; Valas, R.; DuPont, C.L.; McMahon, K.D.; Camacho, A. Metagenomes of Mediterranean coastal lagoons. Sci. Rep. 2012, 2, 490. [Google Scholar] [CrossRef] [PubMed]
- Ventosa, A.; Fernández, A.B.; León, M.J.; Sánchez-Porro, C.; Rodriguez-Valera, F. The Santa Pola saltern as a model for studying the microbiota of hypersaline environments. Extremophiles 2014, 18, 811–24. [Google Scholar] [CrossRef] [PubMed]
- Oren, A. Halophilic microbial communities and their environments. Curr. Opin. Biotechnol. 2015, 33, 119–124. [Google Scholar] [CrossRef]
- Rodriguez-Valera, F.; Rodriguez-Brito, B.; Thingstad, T.F.; Rohwer, F.; Mira, A. Explaining microbial population genomics through phage predation. Nat. Rev. 2009, 7, 828–836. [Google Scholar]
- Montalvo-Rodriguez, R.; Vreeland, R.H.; Aharon, O.; Martin, K.; Lopez-garriga, J.; Chester, W. Halogemetricum borinquense sp. nov., a novel halophilic archaeon from Puerto Rico. Int. J. Syst. Bacteriol. 1998, 48, 1305–1312. [Google Scholar] [CrossRef] [PubMed]
- Montalvo-Rodrıguez, R.; Vreeland, R.H.; Lopez-garriga, J.; Oren, A.; Ventosa, A.; Kamekura, M.; Chester, W. Haloterrigena thermotolerans sp. nov., a halophilic archaeon from Puerto Rico. Int. J. Syst. Evol. Microbiol. 2000, 1065–1071. [Google Scholar]
- Soto-Ramirez, N.; Sanchez-Porro, C.; Rosas, S.; Gonzalez, W.; Quinones, M.; Ventosa, A.; Montalvo-Rodriguez, R. Halomonas avicenniae sp. nov., isolated from the salty leaves of the black mangrove Avicennia germinans in Puerto Rico. Int. J. Syst. Evol. Microbiol. 2007, 57, 900–905. [Google Scholar] [CrossRef]
- Soto-Ramírez, N.; Sánchez-Porro, C.; Rosas-Padilla, S.; Almodóvar, K.; Jiménez, G.; Machado-Rodríguez, M.; Zapata, M.; Ventosa, A.; Montalvo-Rodríguez, R. Halobacillus mangrovi sp. nov., a moderately halophilic bacterium isolated from the black mangrove Avicennia germinans. Int. J. Syst. Evol. Microbiol. 2008, 58, 125–130. [Google Scholar] [CrossRef]
- Sanchez-Porro, C.; de la Haba, R.R.; Soto-Ramirez, N.; Marquez, M.C.; Montalvo-Rodriguez, R.; Ventosa, A. Description of Kushneria aurantia gen. nov., sp. nov., a novel member of the family Halomonadaceae, and a proposal for reclassification of Halomonas marisflavi as Kushneria marisflavi comb. nov., of Halomonas indalinina as Kushneria indalinina comb. nov. and of Halomonas avicenniae as Kushneria avicenniae comb. nov. Int. J. Syst. Evol. Microbiol. 2009, 59, 397–405. [Google Scholar] [PubMed]
- Sánchez-Nieves, R.; Facciotti, M.; Saavedra-Collado, S.; Dávila-Santiago, L.; Rodríguez-Carrero, R.; Montalvo-Rodríguez, R. Draft genome of Haloarcula rubripromontorii strain SL3, a novel halophilic archaeon isolated from the solar salterns of Cabo Rojo, Puerto Rico. Genom. Data 2016, 7, 287–289. [Google Scholar] [CrossRef]
- Sánchez-Nieves, R.; Facciotti, M.T.; Saavedra-Collado, S.; Dávila-Santiago, L.; Rodríguez-Carrero, R.; Montalvo-Rodríguez, R. Draft genome sequence of Halorubrum tropicale strain V5, a novel halophilic archaeon isolated from the solar salterns of Cabo Rojo, Puerto Rico. Genom. Data 2016, 7, 284–286. [Google Scholar] [CrossRef] [PubMed]
- Thomas, T.; Gilbert, J.; Meyer, F. Metagenomics—A guide from sampling to data analysis. Microb. Inform. Exp. 2012, 2, 3. [Google Scholar] [CrossRef] [PubMed]
- Narasingarao, P.; Podell, S.; Ugalde, J.A.; Brochier-Armanet, C.; Emerson, J.B.; Brocks, J.J.; Heidelberg, K.B.; Banfield, J.F.; Allen, E.E. De novo metagenomic assembly reveals abundant novel major lineage of Archaea in hypersaline microbial communities. ISME J. 2012, 6, 81–93. [Google Scholar] [CrossRef] [PubMed]
- Crits-Christoph, A.; Gelsinger, D.R.; Ma, B.; Wierzchos, J.; Ravel, J.; Davila, A.; Casero, M.C.; DiRuggiero, J. Functional interactions of archaea, bacteria and viruses in a hypersaline endolithic community. Environ. Microbiol. 2016, 18, 2064–2077. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-García, C.M. Metagenomic Analysis of Prokaryotic Communities from Hypersaline Environments at Cabo Rojo, Puerto Rico through Pyrosequencing of 16S rRNA Genes. Master’s Thesis, University of Puerto Rico, San Juan, Puerto Rico, 2016. [Google Scholar]
- Martín-Cuadrado, A.-B.; López-García, P.; Alba, J.-C.; Moreira, D.; Monticelli, L.; Strittmatter, A.; Gottschalk, G.; Rodríguez-Valera, F. Metagenomics of the deep Mediterranean, a warm bathypelagic habitat. PLoS ONE 2007, 2, e914. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 1 April 2019).
- Nurk, S.; Meleshko, D.; Korobeynikov, A.; Pevzner, P.A. metaSPAdes: A new versatile metagenomic assembler. Genome Res. 2017, 27, 824–834. [Google Scholar] [CrossRef]
- Meyer, F.; Paarmann, D.; D’Souza, M.; Olson, R.; Glass, E.M.; Kubal, M.; Paczian, T.; Rodriguez, A.; Stevens, R.; Wilke, A.; et al. The metagenomics RAST server—A public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinform. 2008, 9, 386. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Muller, J.; Szklarczyk, D.; Julien, P.; Letunic, I.; Roth, A.; Kuhn, M.; Powell, S.; von Mering, C.; Doerks, T.; Jensen, L.J.; et al. eggNOG v2.0: Extending the evolutionary genealogy of genes with enhanced non-supervised orthologous groups, species and functional annotations. Nucleic Acids Res. 2010, 38, D190–D195. [Google Scholar] [CrossRef]
- Tatusov, R.L.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Kiryutin, B.; Koonin, E.V.; Krylov, D.M.; Mazumder, R.; Mekhedov, S.L.; Nikolskaya, A.N. The COG database: An updated version includes eukaryotes. BMC Bioinform. 2003, 4, 41. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows—Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Kang, D.D.; Froula, J.; Egan, R.; Wang, Z. MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. PeerJ 2015, 3, e1165. [Google Scholar] [CrossRef] [Green Version]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM : Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef]
- Rodriguez-R, L.M.; Gunturu, S.; Harvey, W.T.; Rosselí O-Mora, R.; Tiedje, J.M.; Cole, J.R.; Konstantinidis, K.T. The Microbial Genomes Atlas (MiGA) webserver: Taxonomic and gene diversity analysis of Archaea and Bacteria at the whole genome level. Nucleic Acids Res. 2018, 46, W282–W288. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive tree of life (iTOL) v3: An online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016, 44, W242–W245. [Google Scholar] [CrossRef]
- Overbeek, R.; Olson, R.; Pusch, G.D.; Olsen, G.J.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Parrello, B.; Shukla, M.; et al. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res. 2014, 42, D206–D214. [Google Scholar] [CrossRef]
- Olson, N.D.; Treangen, T.J.; Hill, C.M.; Cepeda-espinoza, V.; Ghurye, J.; Koren, S.; Pop, M. Metagenomic assembly through the lens of validation: Recent advances in assessing and improving the quality of genomes assembled from metagenomes. Brief. Bioinform. 2017, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Mikheenko, A.; Saveliev, V.; Gurevich, A. Genome analysis MetaQUAST: Evaluation of metagenome assemblies. Bioinformatics 2016, 32, 1088–1090. [Google Scholar] [CrossRef]
- Rhodes, M.E.; Oren, A.; House, C.H. The Dynamics and Persistence of Dead Sea Microbial Populations as Shown by High Throughput Sequencing of Ribosomal RNA. Appl. Environ. Microbiol. 2012, 8, 2489–2492. [Google Scholar] [CrossRef]
- Pasic, L.; Bartual, S.G.; Ulrih, N.P.; Grabnar, M.; Velikonja, B.H. Diversity of halophilic archaea in the crystallizers of an Adriatic solar saltern. FEMS Microbiol. Ecol. 2005, 54, 491–498. [Google Scholar] [PubMed] [Green Version]
- Plominsky, A.M.; Henríquez-Castillo, C.; Delherbe, N.; Podell, S.; Ramirez-Flandes, S.; Ugalde, J.A.; Santibañez, J.F.; van den Engh, G.; Hanselmann, K.; Ulloa, O.; et al. Distinctive Archaeal Composition of an Artisanal Crystallizer Pond and Functional Insights Into Salt-Saturated Hypersaline Environment Adaptation. Front. Microbiol. 2018, 9, 1800. [Google Scholar] [CrossRef]
- Podell, S.; Emerson, J.B.; Jones, C.M.; Ugalde, J.A.; Welch, S.; Heidelberg, K.B.; Banfield, J.F.; Allen, E.E. Seasonal fluctuations in ionic concentrations drive microbial succession in a hypersaline lake community. ISME J. 2014, 8, 979–90. [Google Scholar] [CrossRef]
- Peter, H.; Hörtnagl, P.; Reche, I.; Sommaruga, R. Bacterial diversity and composition during rain events with and without Saharan dust influence reaching a high mountain lake in the Alps. Environ. Microbiol. Rep. 2014, 6, 618–624. [Google Scholar] [CrossRef]
- Demergasso, C.; Escudero, L.; Casamayor, E.O.; Chong, G.; Balagué, V.; Pedrós-Alió, C. Novelty and spatio–temporal heterogeneity in the bacterial diversity of hypersaline Lake Tebenquiche (Salar de Atacama). Extremophiles 2008, 12, 491–504. [Google Scholar] [CrossRef]
- Baricz, A.; Coman, C.; Andrei, A.Ş.; Muntean, V.; Keresztes, Z.G.; Păuşan, M.; Alexe, M.; Banciu, H.L. Spatial and temporal distribution of archaeal diversity in meromictic, hypersaline Ocnei Lake (Transylvanian Basin, Romania). Extremophiles 2014, 18, 399–413. [Google Scholar] [CrossRef] [PubMed]
- Atanasova, N.S.; Demina, T.A.; Buivydas, A.; Bamford, D.H.; Oksanen, H.M. Archaeal viruses multiply: Temporal screening in a solar saltern. Viruses 2015, 7, 1902–1926. [Google Scholar] [CrossRef] [PubMed]
- Almeida-Dalmet, S.; Sikaroodi, M.; Gillevet, P.M.; Litchfield, C.D.; Baxter, B.K. Temporal Study of the Microbial Diversity of the North Arm of Great Salt Lake, Utah, U.S. Microorganisms 2015, 3, 310–326. [Google Scholar] [CrossRef]
- Javor, B.J. Hypersaline Environments: Microbiology and Biogeochemistry; Springer Science & Business Media: Berlin, Germany, 2012; ISBN 3642743706. [Google Scholar]
- Joint, I.; Henriksen, P.; Garde, K.; Riemann, B. Primary production, nutrient assimilation and microzooplankton grazing along a hypersaline gradient. FEMS Microbiol. Ecol. 2002, 39, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Oren, A. The ecology of Dunaliella in high-salt environments. J. Biol. Res. 2014, 21, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ríos, A.d.l.; Valea, S.; Ascaso, C.; Davila, A.F.; Kastovsky, J.; McKay, C.P.; Wierzchos, J. Comparative analysis of the microbial communities inhabiting halite evaporites of the Atacama Desert. Int. Microbiol. 2010, 13, 79–89. [Google Scholar]
- Payler, S.J.; Biddle, J.F.; Sherwood Lollar, B.; Fox-Powell, M.G.; Edwards, T.; Ngwenya, B.T.; Paling, S.M.; Cockell, C.S. An Ionic Limit to Life in the Deep Subsurface. Front. Microbiol. 2019, 10, 426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabello, P.; Roldan, M.D.; Moreno-Vivian, C. Nitrate reduction and the nitrogen cycle in archaea. Microbiology 2004, 150, 3527–3546. [Google Scholar] [CrossRef]
- Offre, P.; Spang, A.; Schleper, C. Archaea in Biogeochemical Cycles. Annu. Rev. Microbiol. 2013, 16, 437–457. [Google Scholar] [CrossRef] [PubMed]
- Stahl, D.A.; Torre, R. De Physiology and Diversity of Ammonia-Oxidizing Archaea. Annu. Rev. Microbiol. 2012, 66, 83–101. [Google Scholar] [CrossRef]
- Könneke, M.; Bernhard, A.E.; José, R.; Walker, C.B.; Waterbury, J.B.; Stahl, D.A. Isolation of an autotrophic ammonia-oxidizing marine archaeon. Nature 2005, 437, 543. [Google Scholar] [CrossRef] [PubMed]
- Walker, C.B.; De La Torre, J.R.; Klotz, M.G.; Urakawa, H.; Pinel, N.; Arp, D.J.; Brochier-Armanet, C.; Chain, P.S.G.; Chan, P.P.; Gollabgir, A. Nitrosopumilus maritimus genome reveals unique mechanisms for nitrification and autotrophy in globally distributed marine crenarchaea. Proc. Natl. Acad. Sci. USA 2010, 107, 8818–8823. [Google Scholar] [CrossRef] [PubMed]
- Oren, A. Bioenergetic aspects of halophilism. Microbiol. Mol. Biol. Rev. 1999, 63, 334–348. [Google Scholar] [PubMed]
- Muyzer, G.; Stams, A.J.M. The ecology and biotechnology of sulphate-reducing bacteria. Nat. Rev. 2008, 6, 441–454. [Google Scholar] [CrossRef] [PubMed]
- Barton, L.L.; Fauque, G.D. Biochemistry, physiology and biotechnology of sulfate-reducing bacteria. Adv. Appl. Microbiol. 2009, 68, 41–98. [Google Scholar] [PubMed]
- Tourova, T.P.; Kovaleva, O.L.; Sorokin, D.Y.; Muyzer, G. Ribulose-1, 5-bisphosphate carboxylase/oxygenase genes as a functional marker for chemolithoautotrophic halophilic sulfur-oxidizing bacteria in hypersaline habitats. Microbiology 2010, 156, 2016–2025. [Google Scholar] [CrossRef]
- Segerer, A.; Neuner, A.; Kristjansson, J.K.; Stetter, K.O. Acidianus infernus gen. nov., sp. nov., and Acidianus brierleyi comb. nov.: Facultatively aerobic, extremely acidophilic thermophilic sulfur-metabolizing archaebacteria. Int. J. Syst. Evol. Microbiol. 1986, 36, 559–564. [Google Scholar] [CrossRef]
- Hafenbradl, D.; Keller, M.; Dirmeier, R.; Rachel, R.; Roßnagel, P.; Burggraf, S.; Huber, H.; Stetter, K.O. Ferroglobus placidus gen. nov., sp. nov., a novel hyperthermophilic archaeum that oxidizes Fe2+ at neutral pH under anoxic conditions. Arch. Microbiol. 1996, 2, 308–314. [Google Scholar] [CrossRef]
- Rodriguez-R, L.M.; Konstantinidis, K.T. Bypassing Cultivation to Identify Bacterial Species Culture-independent genomic approaches identify credibly distinct clusters, avoid cultivation bias, and provide true insights into microbial species. Microbe 2014, 9, 111–118. [Google Scholar]
- Arahal, D.R. Whole-Genome Analyses: Average Nucleotide Identity, 1st ed.; Elsevier Ltd.: Amsterdam, The Netherlands, 2014; Volume 41. [Google Scholar]
- Goris, J.; Konstantinidis, K.T.; Klappenbach, J.A.; Coenye, T.; Vandamme, P.; Tiedje, J.M. DNA—DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 2007, 57, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Konstantinidis, K.T.; Tiedje, J.M. Towards a Genome-Based Taxonomy for Prokaryotes. J. Bacteriol. 2005, 187, 6258–6264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richter, M.; Rossello, R. Shifting the genomic gold standard for the prokaryotic species definition. PNAS 2009, 106, 19126–19131. [Google Scholar] [CrossRef] [Green Version]
- Luo, C.; Rodriguez-R, L.M.; Konstantinidis, K.T. A User’s Guide to Quantitative and Comparative Analysis of Metagenomic Datasets, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2013; Volume 531, ISBN 9780124078635. [Google Scholar]
- Oren, A.; Elevi, R.; Watanabe, S.; Ihara, K.; Corcelli, A. Halomicrobium mukohataei gen. nov., comb. nov., and emended description of Halomicrobium mukohataei. Int. J. Syst. Evol. Microbiol. 2002, 52, 1831–1835. [Google Scholar] [PubMed]
- Nishimura, T.; Vertès, A.A.; Shinoda, Y. Anaerobic growth of Corynebacterium glutamicum using nitrate as a terminal electron acceptor. Appl. Microbiol. Biotechnol. 2007, 75, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Bolhuis, H.; Palm, P.; Wende, A.; Falb, M.; Rampp, M.; Rodriguez-valera, F.; Pfeiffer, F.; Oesterhelt, D. The genome of the square archaeon Haloquadratum walsbyi: Life at the limits of water activity. BMC Genom. 2006, 7, 1–12. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Number of Reads | GC Content (%) | Read Length (bp) | Number of contigs | N50 (bp) | Longest Contig (bp) |
|---|---|---|---|---|---|---|
| MFF1 | 29,432,758 | 56 | 35–251 | 318,469 | 3888 | 619,112 |
| MFF2 | 41,746,817 | 57 | 35–151 | 420,402 | 4748 | 469,957 |
| MFF3 | 40,634,465 | 58 | 35–151 | 379,415 | 4854 | 388,630 |
| MFF1 | MFF2 | MFF3 | |||
|---|---|---|---|---|---|
| Genus | Abundance | Genus | Abundance | Genus | Abundance |
| Haloquadratum | 53.77% | Haloquadratum | 69.76% | Haloquadratum | 62.47% |
| Salinibacter | 16.02% | Salinibacter | 12.99% | Salinibacter | 21.17% |
| Halorubrum | 8.65% | Halorubrum | 7.55% | Halorubrum | 4.33% |
| Unclassified* | 2.74% | Halococcus | 1.99% | Haloplanus | 4.12% |
| Haloplanus | 2.39% | Natronomonas | 1.39% | Halococcus | 2.41% |
| Haloarcula | 1.84% | Halomicrobium | 1.08% | Haloterrigena | 2.01% |
| Pseudomonas | 1.74% | Haloterrigena | 1.07% | Natrinema | 1.08% |
| Halococcus | 1.12% | Haloplanus | 1.05% | Natronomonas | 0.72% |
| Natronomonas | 1.10% | Haloferax | 0.45% | Halovivax | 0.58% |
| Haloferax | 0.94% | Haloarcula | 0.43% | Halobaculum | 0.51% |
| Halovivax | 0.66% | Halobaculum | 0.41% | Haloferax | 0.35% |
| Dyella | 0.61% | Halobacterium | 0.41% | Halomicrobium | 0.10% |
| Ruminococcus | 0.46% | Halogeometricum | 0.37% | Haloarcula | 0.10% |
| Total sequences | 19,228 | 37,156 | 31,202 | ||
| Bin Name | Completeness (%) | Contamination (%) | GC Content (%) | Number of Contigs | N50 (bp) | Genome Size (Mb) | Predicted Proteins |
|---|---|---|---|---|---|---|---|
| RC33 | 94.40 | 2.40 | 66.22 | 11 | 392,903 | 1.5 | 1576 |
| RC24 | 80.20 | 1.80 | 52.79 | 28 | 125,394 | 1.8 | 1533 |
| RC39 | 96.00 | 3,20 | 68.18 | 75 | 54,228 | 2.4 | 2502 |
| RC20 | 88.66 | 3.20 | 50.25 | 273 | 24,930 | 4.3 | 4449 |
| Bin | Closest Relative | AAI | Fraction of Proteins Shared |
|---|---|---|---|
| RC33 | Natronomonas moolapensis | 59.74% | 81.85% |
| RC24 | Pontibacter korlensis | 43.31% | 66.02% |
| RC39 | Halomicrobium mukohataei | 62.81% | 68.27% |
| RC20 | Haloquadratum walsbyi | 65.83% | 73.46% |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Couto-Rodríguez, R.L.; Montalvo-Rodríguez, R. Temporal Analysis of the Microbial Community from the Crystallizer Ponds in Cabo Rojo, Puerto Rico, Using Metagenomics. Genes 2019, 10, 422. https://doi.org/10.3390/genes10060422
Couto-Rodríguez RL, Montalvo-Rodríguez R. Temporal Analysis of the Microbial Community from the Crystallizer Ponds in Cabo Rojo, Puerto Rico, Using Metagenomics. Genes. 2019; 10(6):422. https://doi.org/10.3390/genes10060422
Chicago/Turabian StyleCouto-Rodríguez, Ricardo L., and Rafael Montalvo-Rodríguez. 2019. "Temporal Analysis of the Microbial Community from the Crystallizer Ponds in Cabo Rojo, Puerto Rico, Using Metagenomics" Genes 10, no. 6: 422. https://doi.org/10.3390/genes10060422
APA StyleCouto-Rodríguez, R. L., & Montalvo-Rodríguez, R. (2019). Temporal Analysis of the Microbial Community from the Crystallizer Ponds in Cabo Rojo, Puerto Rico, Using Metagenomics. Genes, 10(6), 422. https://doi.org/10.3390/genes10060422