Genomic Evidence of Recombination in the Basidiomycete Wallemia mellicola


 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Culture, Medium, Growth Conditions and DNA Isolation
2.2. Genome Sequencing
2.3. Variant Calling
2.4. Assembly and Annotation
2.5. Variant-Based Analysis
2.6. Phylogenetic Analysis
2.7. Core Genome, GO Enrichment
2.8. Mating-Type Loci
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zajc, J.; Gunde-Cimerman, N. The Genus Wallemia—From Contamination of Food to Health Threat. Microorganisms 2018, 6, 46. [Google Scholar] [CrossRef] [PubMed]
- Johan-Olsen, O. Om Sop på Klipfisk den Såkaldte Mid; Dybwad: Christiania, Norway, 1887. [Google Scholar]
- Zalar, P.; Sybren de Hoog, G.; Schroers, H.-J.; Frank, J.M.; Gunde-Cimerman, N. Taxonomy and phylogeny of the xerophilic genus Wallemia (Wallemiomycetes and Wallemiales, cl. et ord. nov.). Antonie Van Leeuwenhoek 2005, 87, 311–328. [Google Scholar] [CrossRef] [PubMed]
- Jančič, S.; Nguyen, H.D.T.; Frisvad, J.C.; Zalar, P.; Schroers, H.-J.; Seifert, K.A.; Gunde-Cimerman, N. A Taxonomic Revision of the Wallemia sebi Species Complex. PLoS ONE 2015, 10, e0125933. [Google Scholar] [CrossRef] [PubMed]
- Jančič, S.; Zalar, P.; Kocev, D.; Schroers, H.J.; Džeroski, S.; Gunde-Cimerman, N. Halophily reloaded: New insights into the extremophilic life-style of Wallemia with the description of Wallemia hederae sp. nov. Fungal Divers. 2016, 76, 97–118. [Google Scholar] [CrossRef]
- Díaz-Valderrama, J.R.; Nguyen, H.D.T.; Aime, M.C. Wallemia peruviensis sp. nov., a new xerophilic fungus from an agricultural setting in South America. Extremophiles 2017, 21, 1017–1025. [Google Scholar] [CrossRef]
- Matheny, P.B.; Gossmann, J.A.; Zalar, P.; Kumar, T.K.A.; Hibbett, D.S. Resolving the phylogenetic position of the Wallemiomycetes: An enigmatic major lineage of Basidiomycota. Can. J. Bot. 2006, 84, 1794–1805. [Google Scholar] [CrossRef]
- Padamsee, M.; Kumar, T.K.A.; Riley, R.; Binder, M.; Boyd, A.; Calvo, A.M.; Furukawa, K.; Hesse, C.; Hohmann, S.; James, T.Y.; et al. The genome of the xerotolerant mold Wallemia sebi reveals adaptations to osmotic stress and suggests cryptic sexual reproduction. Fungal Genet. Biol. 2012, 49, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Zajc, J.; Liu, Y.; Dai, W.; Yang, Z.; Hu, J.; Gostinčar, C.; Gunde-Cimerman, N. Genome and transcriptome sequencing of the halophilic fungus Wallemia ichthyophaga: Haloadaptations present and absent. BMC Genom. 2013, 14, 617. [Google Scholar] [CrossRef]
- Zhao, R.L.; Li, G.J.; Sánchez-Ramírez, S.; Stata, M.; Yang, Z.L.; Wu, G.; Dai, Y.C.; He, S.H.; Cui, B.K.; Zhou, J.L.; et al. A six-gene phylogenetic overview of Basidiomycota and allied phyla with estimated divergence times of higher taxa and a phyloproteomics perspective. Fungal Divers. 2017, 84, 43–74. [Google Scholar] [CrossRef]
- Pitt, J.I.; Hocking, A.D. Fungi and Food Spoilage, 2nd ed.; Aspen Publishers, Inc.: Gaithersburg, MD, USA, 1999; ISBN 978-0-387-92206-5. [Google Scholar]
- Samson, R.A.; Hoekstra, E.S.; Frisvad, J.C.; Filtenborg, O. Introduction to Food- and Airborne Fungi, 6th ed.; Centraalbureau voor Schimmelcultures: Baarn, The Netherlands, 2002. [Google Scholar]
- Takahashi, T. Airborne fungal colony-forming units in outdoor and indoor environments in Yokohama, Japan. Mycopathologia 1997, 139, 23–33. [Google Scholar] [CrossRef]
- Fröhlich-Nowoisky, J.; Pickersgill, D.A.; Despres, V.R.; Poschl, U. High diversity of fungi in air particulate matter. Proc. Natl. Acad. Sci. USA 2009, 106, 12814–12819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubert, J.; Nesvorna, M.; Kopecky, J.; Erban, T.; Klimov, P. Population and Culture Age Influence the Microbiome Profiles of House Dust Mites. Microb. Ecol. 2019, 77, 1048–1066. [Google Scholar] [CrossRef] [PubMed]
- Zajc, J.; Kogej, T.; Ramos, J.; Galinski, E.A.; Gunde-Cimerman, N. The osmoadaptation strategy of the most halophilic fungus Wallemia ichthyophaga, growing optimally at salinities above 15% NaCl. Appl. Environ. Microbiol. 2014, 80, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Jančič, S.; Frisvad, J.C.; Kocev, D.; Gostinčar, C.; Džeroski, S.; Gunde-Cimerman, N. Production of secondary metabolites in extreme environments: Food- and airborne Wallemia spp. produce toxic metabolites at hypersaline conditions. PLoS ONE 2016, 11, e0169116. [Google Scholar] [CrossRef] [PubMed]
- Moss, M.O. Recent studies of mycotoxins. J. Appl. Microbiol. 1998, 84, 62–76. [Google Scholar] [CrossRef]
- Guarro, J.; Gugnani, H.C.; Sood, N.; Batra, R.; Mayayo, E.; Gene, J.; Kakkar, S. Subcutaneous phaeohyphomycosis caused by Wallemia sebi in an immunocompetent host. J. Clin. Microbiol. 2008, 46, 1129–1131. [Google Scholar] [CrossRef]
- Sakamoto, T.; Urisu, A.; Yamada, M.; Matsuda, Y.; Tanaka, K.; Torii, S. Studies on the Osmophilic Fungus Wallemia sebi as an Allergen Evaluated by Skin Prick Test and Radioallergosorbent Test. Int. Arch. Allergy Immunol. 1989, 90, 368–372. [Google Scholar] [CrossRef]
- Vesper, S.J.; McKinstry, C.; Yang, C.; Haugland, R.A.; Kercsmar, C.M.; Yike, I.; Schluchter, M.D.; Kirchner, H.L.; Sobolewski, J.; Allan, T.M.; et al. Specific molds associated with asthma in water-damaged homes. J. Occup. Environ. Med. 2006, 48, 852–858. [Google Scholar] [CrossRef]
- Wheeler, M.L.; Limon, J.J.; Bar, A.S.; Leal, C.A.; Gargus, M.; Tang, J.; Brown, J.; Funari, V.A.; Wang, H.L.; Crother, T.R.; et al. Immunological Consequences of Intestinal Fungal Dysbiosis. Cell Host Microbe 2016, 19, 865–873. [Google Scholar] [CrossRef] [Green Version]
- Skalski, J.H.; Limon, J.J.; Sharma, P.; Gargus, M.D.; Nguyen, C.; Tang, J.; Coelho, A.L.; Hogaboam, C.M.; Crother, T.R.; Underhill, D.M. Expansion of commensal fungus Wallemia mellicola in the gastrointestinal mycobiota enhances the severity of allergic airway disease in mice. PLoS Pathog. 2018, 14, e1007260. [Google Scholar] [CrossRef]
- Gostinčar, C.; Stajich, J.E.; Zupančič, J.; Zalar, P.; Gunde-Cimerman, N. Genomic evidence for intraspecific hybridization in a clonal and extremely halotolerant yeast. BMC Genom. 2018, 19, 364. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Zhong, H.; Lin, Y.; Chen, B.; Han, M.; Ren, H.; Lu, H.; Luber, J.M.; Xia, M.; Li, W.; et al. Assessment of the cPAS-based BGISEQ-500 platform for metagenomic sequencing. Gigascience 2018, 7, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alkan, C.; Coe, B.P.; Eichler, E.E. GATK toolkit. Nat. Rev. Genet. 2011, 12, 363–376. [Google Scholar] [CrossRef]
- Peng, Y.; Leung, H.C.M.; Yiu, S.M.; Chin, F.Y.L. IDBA-UD: A de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 2012, 28, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Campbell, M.S.; Holt, C.; Moore, B.; Yandell, M. Genome Annotation and Curation Using MAKER and MAKER-P. Curr. Protoc. Bioinform. 2014, 48, 1–39. [Google Scholar] [CrossRef]
- Korf, I. Gene finding in novel genomes. BMC Bioinform. 2004, 5, 59. [Google Scholar] [CrossRef] [PubMed]
- Stanke, M.; Morgenstern, B. AUGUSTUS: A web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res. 2005, 33, 465–467. [Google Scholar] [CrossRef] [PubMed]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef]
- Kriventseva, E.V.; Tegenfeldt, F.; Petty, T.J.; Waterhouse, R.M.; Simão, F.A.; Pozdnyakov, I.A.; Ioannidis, P.; Zdobnov, E.M. OrthoDB v8: Update of the hierarchical catalog of orthologs and the underlying free software. Nucleic Acids Res. 2015, 43, 250–256. [Google Scholar] [CrossRef]
- Geib, S.M.; Hall, B.; Derego, T.; Bremer, F.T.; Cannoles, K.; Sim, S.B. Genome Annotation Generator: A simple tool for generating and correcting WGS annotation tables for NCBI submission. Gigascience 2018, 7, 4. [Google Scholar] [CrossRef] [PubMed]
- Jombart, T.; Ahmed, I. adegenet 1.3-1: New tools for the analysis of genome-wide SNP data. Bioinformatics 2011, 27, 3070–3071. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. ggplot2; Springer New York: New York, NY, USA, 2009; ISBN 978-0-387-98140-6. [Google Scholar]
- R Development Core Team R: A Language and Environment for Statistical Computing. 2019. Available online: ftp://ftp.uvigo.es/CRAN/web/packages/dplR/vignettes/intro-dplR.pdf (accessed on 23 March 2019).
- Katoh, K.; Toh, H. Recent developments in the MAFFT multiple sequence alignment program. Brief. Bioinform. 2008, 9, 286–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talavera, G.; Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef]
- Hasegawa, M.; Kishino, H.; Yano, T. aki Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J. Mol. Evol. 1985, 22, 160–174. [Google Scholar] [CrossRef]
- Bouckaert, R.R. DensiTree: Making sense of sets of phylogenetic trees. Bioinformatics 2010, 26, 1372–1373. [Google Scholar] [CrossRef]
- Revell, L.J. phytools: An R package for phylogenetic comparative biology (and other things). Methods Ecol. Evol. 2012, 3, 217–223. [Google Scholar] [CrossRef]
- Vinuesa, P.; Contreras-Moreira, B. Robust identification of orthologues and paralogues for microbial pan-genomics using GET_HOMOLOGUES: A case study of pIncA/C plasmids. Methods Mol. Biol. 2015, 1231, 203–232. [Google Scholar] [CrossRef] [PubMed]
- Thomas, P.D. PANTHER: A Library of Protein Families and Subfamilies Indexed by Function. Genome Res. 2003, 13, 2129–2141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurtz, S.; Phillippy, A.; Delcher, A.L.; Smoot, M.; Shumway, M.; Antonescu, C.; Salzberg, S.L. Versatile and open software for comparing large genomes. Genome Biol. 2004, 5, 2. [Google Scholar] [CrossRef] [PubMed]
- Gostinčar, C.; Grube, M.; De Hoog, S.; Zalar, P.; Gunde-Cimerman, N. Extremotolerance in fungi: Evolution on the edge. FEMS Microbiol. Ecol. 2010, 71, 2–11. [Google Scholar] [CrossRef]
- Gostinčar, C.; Grube, M.; Gunde-Cimerman, N. Evolution of fungal pathogens in domestic environments? Fungal Biol. 2011, 115, 1008–1018. [Google Scholar] [CrossRef] [PubMed]
- Gostinčar, C.; Gunde-Cimerman, N.; Grube, M. 10 Polyextremotolerance as the fungal answer to changing environments. In Microbial Evolution under Extreme Conditions; Bakermans, C., Ed.; DE GRUYTER: Berlin/München,Germany; Boston, MA, USA, 2015; pp. 185–208. ISBN 9783110335064. [Google Scholar]
- Silva, D.N.; Várzea, V.; Paulo, O.S.; Batista, D. Population genomic footprints of host adaptation, introgression and recombination in coffee leaf rust. Mol. Plant Pathol. 2018, 19, 1742–1753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhami, M.K.; Hartwig, T.; Letten, A.D.; Banf, M.; Fukami, T. Genomic diversity of a nectar yeast clusters into metabolically, but not geographically, distinct lineages. Mol. Ecol. 2018, 27, 2067–2076. [Google Scholar] [CrossRef] [PubMed]
- Branco, S.; Gladieux, P.; Ellison, C.E.; Kuo, A.; LaButti, K.; Lipzen, A.; Grigoriev, I.V.; Liao, H.L.; Vilgalys, R.; Peay, K.G.; et al. Genetic isolation between two recently diverged populations of a symbiotic fungus. Mol. Ecol. 2015, 24, 2747–2758. [Google Scholar] [CrossRef]
- Ellison, C.E.; Hall, C.; Kowbel, D.; Welch, J.; Brem, R.B.; Glass, N.L.; Taylor, J.W. Population genomics and local adaptation in wild isolates of a model microbial eukaryote. Proc. Natl. Acad. Sci. USA 2011, 108, 2831–2836. [Google Scholar] [CrossRef] [Green Version]
- Gregory, T.R.; Nicol, J.A.; Tamm, H.; Kullman, B.; Kullman, K.; Leitch, I.J.; Murray, B.G.; Kapraun, D.F.; Greilhuber, J.; Bennett, M.D. Eukaryotic genome size databases. Nucleic Acids Res. 2007, 35, D332–D338. [Google Scholar] [CrossRef]
- Mohanta, T.K.; Bae, H. The diversity of fungal genome. Biol. Proced. Online 2015, 17, 8. [Google Scholar] [CrossRef] [PubMed]
- Peter, J.; De Chiara, M.; Friedrich, A.; Yue, J.-X.; Pflieger, D.; Bergström, A.; Sigwalt, A.; Barre, B.; Freel, K.; Llored, A.; et al. Genome evolution across 1,011 Saccharomyces cerevisiae isolates. Nature 2018, 556, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Pomraning, K.R.; Smith, K.M.; Freitag, M. Bulk Segregant Analysis Followed by High-Throughput Sequencing Reveals the Neurospora Cell Cycle Gene, ndc-1, To Be Allelic with the Gene for Ornithine Decarboxylase, spe-1. Eukaryot. Cell 2011, 10, 724–733. [Google Scholar] [CrossRef] [PubMed]
- Carreté, L.; Ksiezopolska, E.; Pegueroles, C.; Gómez-Molero, E.; Saus, E.; Iraola-Guzmán, S.; Loska, D.; Bader, O.; Fairhead, C.; Gabaldón, T. Patterns of Genomic Variation in the Opportunistic Pathogen Candida glabrata Suggest the Existence of Mating and a Secondary Association with Humans. Curr. Biol. 2018, 28, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Branco, S.; Bi, K.; Liao, H.-L.; Gladieux, P.; Badouin, H.; Ellison, C.E.; Nguyen, N.H.; Vilgalys, R.; Peay, K.G.; Taylor, J.W.; et al. Continental-level population differentiation and environmental adaptation in the mushroom Suillus brevipes. Mol. Ecol. 2017, 26, 2063–2076. [Google Scholar] [CrossRef] [PubMed]
- Desjardins, C.A.; Giamberardino, C.; Sykes, S.M.; Yu, C.-H.; Tenor, J.L.; Chen, Y.; Yang, T.; Jones, A.M.; Sun, S.; Haverkamp, M.R.; et al. Population genomics and the evolution of virulence in the fungal pathogen Cryptococcus neoformans. Genome Res. 2017, 27, 1207–1219. [Google Scholar] [CrossRef] [PubMed]
- Savary, R.; Masclaux, F.G.; Wyss, T.; Droh, G.; Cruz Corella, J.; Machado, A.P.; Morton, J.B.; Sanders, I.R. A population genomics approach shows widespread geographical distribution of cryptic genomic forms of the symbiotic fungus Rhizophagus irregularis. ISME J. 2018, 12, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Tibayrenc, M.; Ayala, F.J. Reproductive clonality of pathogens: A perspective on pathogenic viruses, bacteria, fungi, and parasitic protozoa. Proc. Natl. Acad. Sci. USA 2012, 109, 3305–3313. [Google Scholar] [CrossRef]
- Taylor, J.W.; Hann-Soden, C.; Branco, S.; Sylvain, I.; Ellison, C.E. Clonal reproduction in fungi. Proc. Natl. Acad. Sci. USA 2015, 112, 8901–8908. [Google Scholar] [CrossRef] [Green Version]
- Nieuwenhuis, B.P.S.; James, T.Y. The frequency of sex in fungi. Philos. Trans. R. Soc. B Biol. Sci. 2016, 371, 20150540. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
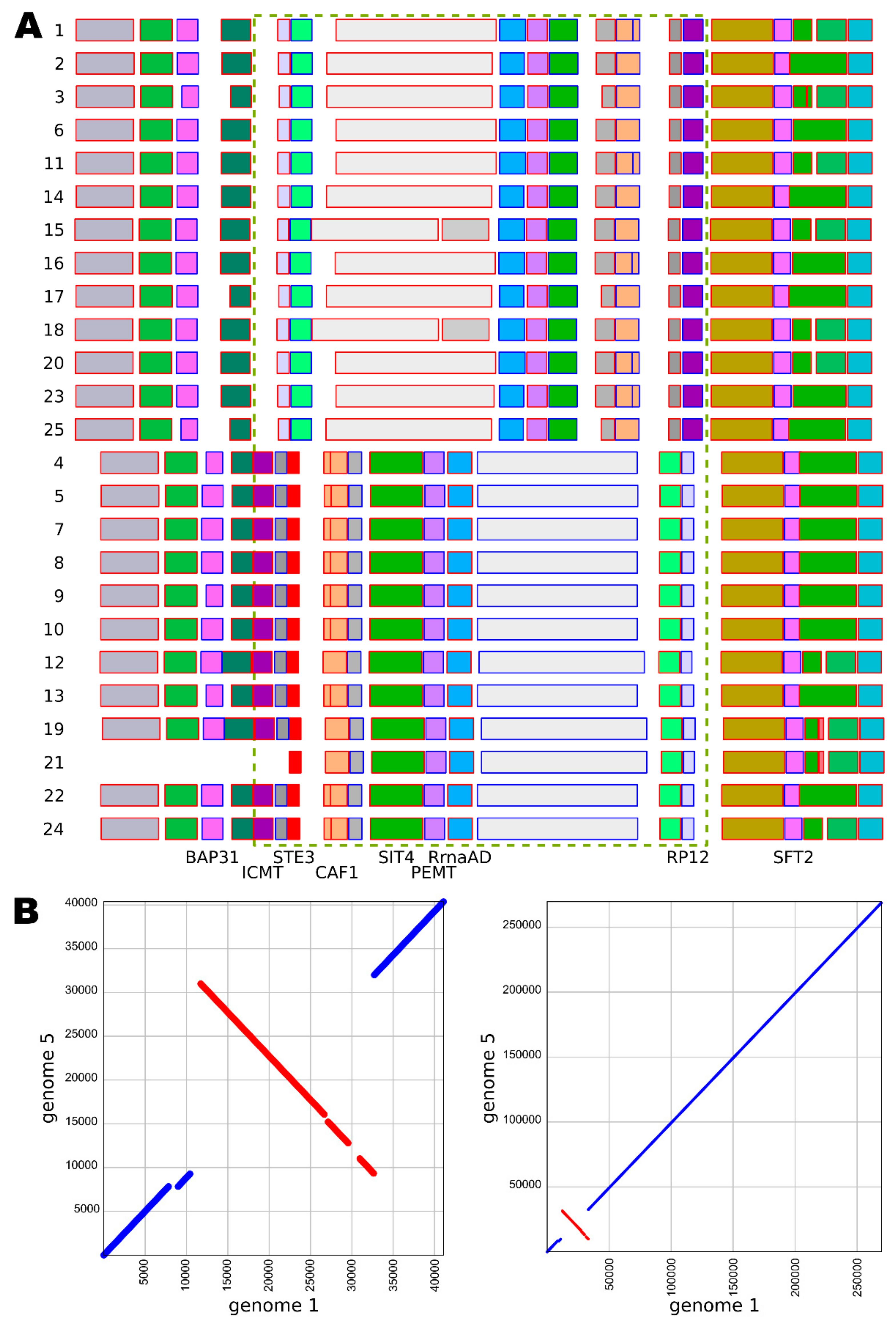{kind=link}
| Culture Collection Strain Number * | Number in This Study | Isolation Habitat | Sampling Site Location |
|---|---|---|---|
| EXF-277 | 1 | hypersaline saltern water | Spain |
| EXF-757 | 2 | hypersaline saltern water | Dominican Republic |
| EXF-1274 (CBS 110588) | 3 | peanuts | Indonesia |
| EXF-1277 (CBS 110589) | 4 | Channa striata dried salted fish | Indonesia |
| EXF-1279 (CBS 110593) | 5 | straw hat | Philippines |
| EXF-5677 | 6 | air | Slovenia |
| EXF-5829 | 7 | chocolate | Slovenia |
| EXF-6156 (UAMH 2651) | 8 | moldy white bread | United Kingdom |
| EXF-6157 (UAMH 2757) | 9 | soil | Canada |
| EXF-6158 (UAMH 6689) | 10 | maple syrup | Canada |
| EXF-8738 | 11 | house dust | Uruguay |
| EXF-8740 | 12 | house dust | Micronesia |
| EXF-8747 | 13 | house dust | Indonesia |
| EXF-8749 | 14 | house dust | Thailand |
| EXF-8757 | 15 | house dust | Mexico |
| EXF-10633 | 16 | dry common fig | Slovenia |
| EXF-483 | 17 | hypersaline saltern water | Spain |
| EXF-1262 (CBS 213.34) | 18 | Unknown | Italy |
| EXF-1443 (IBT 19078) | 19 | Unknown | Denmark |
| EXF-5828 | 20 | chocolate | Slovenia |
| EXF-5830 | 21 | chocolate | Slovenia |
| EXF-5922 | 22 | chocolate | Slovenia |
| EXF-6152 (MUCL 45613) | 23 | forest plant (Clusia rosea) | Cuba |
| EXF-6151 (MUCL 45615) | 24 | forest plant (Verbena officinalis) | Cuba |
| EXF-8741 | 25 | house dust | Micronesia |
| Statistic * | Minimum ** | Mean ** | Maximum ** | Standard Deviation** |
|---|---|---|---|---|
| Coverage | 194 | 318 | 558 | 92 |
| Genome assembly size (Mbp) | 9.68 | 9.75 | 9.95 | 0.05 |
| Number of contigs | 202 | 239 | 422 | 43 |
| Contig N50 | 115375 | 144560 | 170540 | 13888 |
| GC content (%) | 39.91% | 39.95% | 39.97% | 0.01% |
| CDS total length (Mbp) | 6.44 | 6.50 | 6.53 | 0.02 |
| CDS total length (% of genome) | 66.45% | 66.64% | 67.08% | 0.32% |
| Gene models (n) | 4317 | 4475 | 4509 | 37 |
| CDS average length (bp) | 1438 | 1453 | 1512 | 13 |
| Exons per gene (average) | 3.98 | 4.02 | 4.17 | 0.04 |
| Intron average length (bp) | 63 | 64 | 66 | 0.57 |
| Complete BUSCOs | 87.40% | 88.18% | 89.80% | 0.50% |
| Complete and single-copy BUSCOs | 85.90% | 87.91% | 88.60% | 0.56% |
| Complete and duplicated BUSCOs | 0.10% | 0.27% | 3.90% | 0.76% |
| Fragmented BUSCOs | 5.50% | 5.94% | 6.50% | 0.31% |
| Missing BUSCOs | 4.60% | 5.89% | 6.50% | 0.38% |
| SNP density | 0.41% | 0.52% | 0.60% | 0.04% |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, X.; Gostinčar, C.; Fang, C.; Zajc, J.; Hou, Y.; Song, Z.; Gunde-Cimerman, N. Genomic Evidence of Recombination in the Basidiomycete Wallemia mellicola. Genes 2019, 10, 427. https://doi.org/10.3390/genes10060427
Sun X, Gostinčar C, Fang C, Zajc J, Hou Y, Song Z, Gunde-Cimerman N. Genomic Evidence of Recombination in the Basidiomycete Wallemia mellicola. Genes. 2019; 10(6):427. https://doi.org/10.3390/genes10060427
Chicago/Turabian StyleSun, Xiaohuan, Cene Gostinčar, Chao Fang, Janja Zajc, Yong Hou, Zewei Song, and Nina Gunde-Cimerman. 2019. "Genomic Evidence of Recombination in the Basidiomycete Wallemia mellicola" Genes 10, no. 6: 427. https://doi.org/10.3390/genes10060427
APA StyleSun, X., Gostinčar, C., Fang, C., Zajc, J., Hou, Y., Song, Z., & Gunde-Cimerman, N. (2019). Genomic Evidence of Recombination in the Basidiomycete Wallemia mellicola. Genes, 10(6), 427. https://doi.org/10.3390/genes10060427