Defining the Clinical, Molecular and Ultrastructural Characteristics in Occipital Horn Syndrome: Two New Cases and Review of the Literature

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients
2.1.1. Clinical Reports
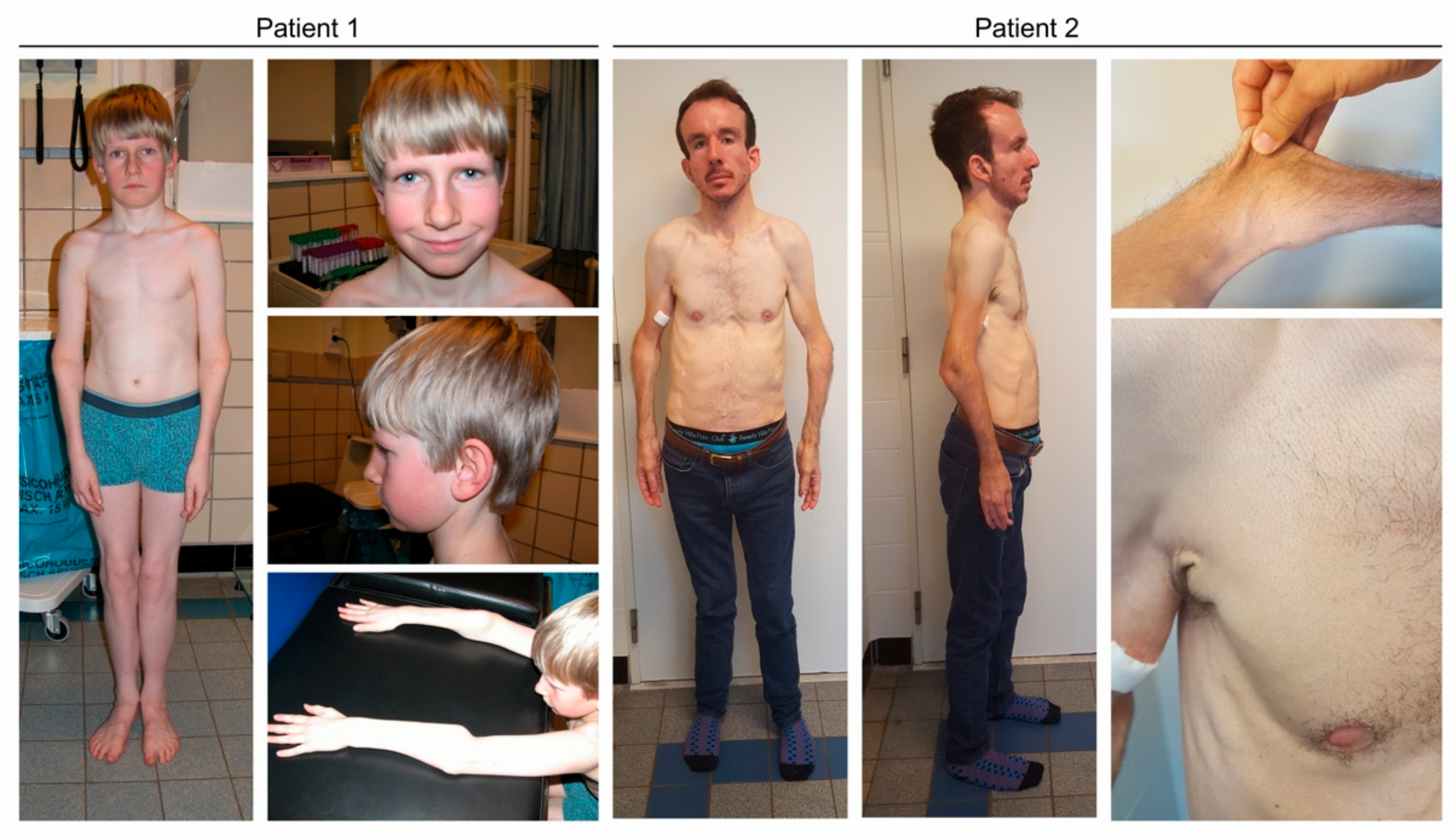
Case 1
Case 2
2.2. Molecular Analysis
2.3. Transmission Electron Microscopy (TEM)
2.4. Collagen Biochemical Analysis
3. Results
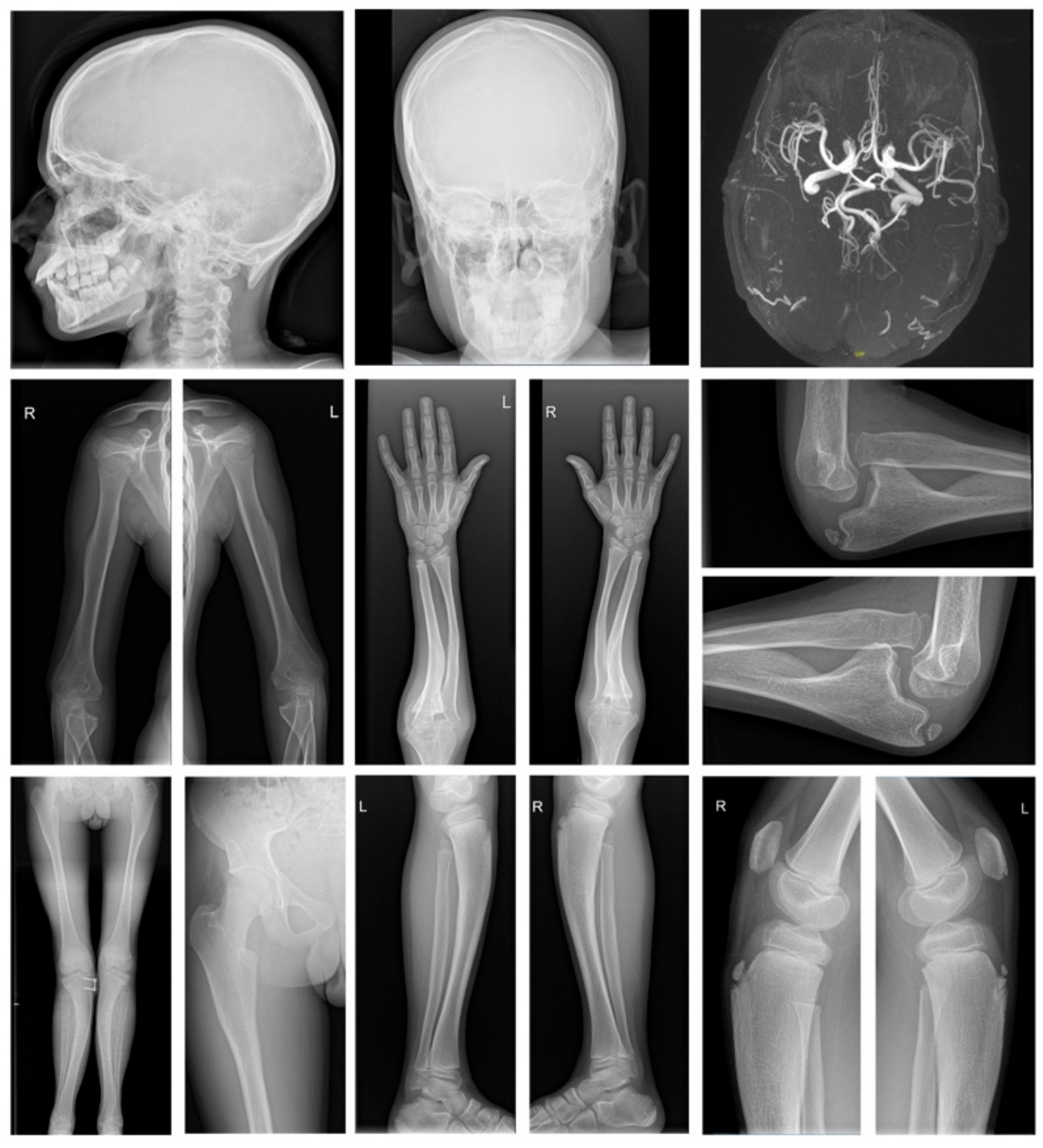
3.1. Clinical Characteristics
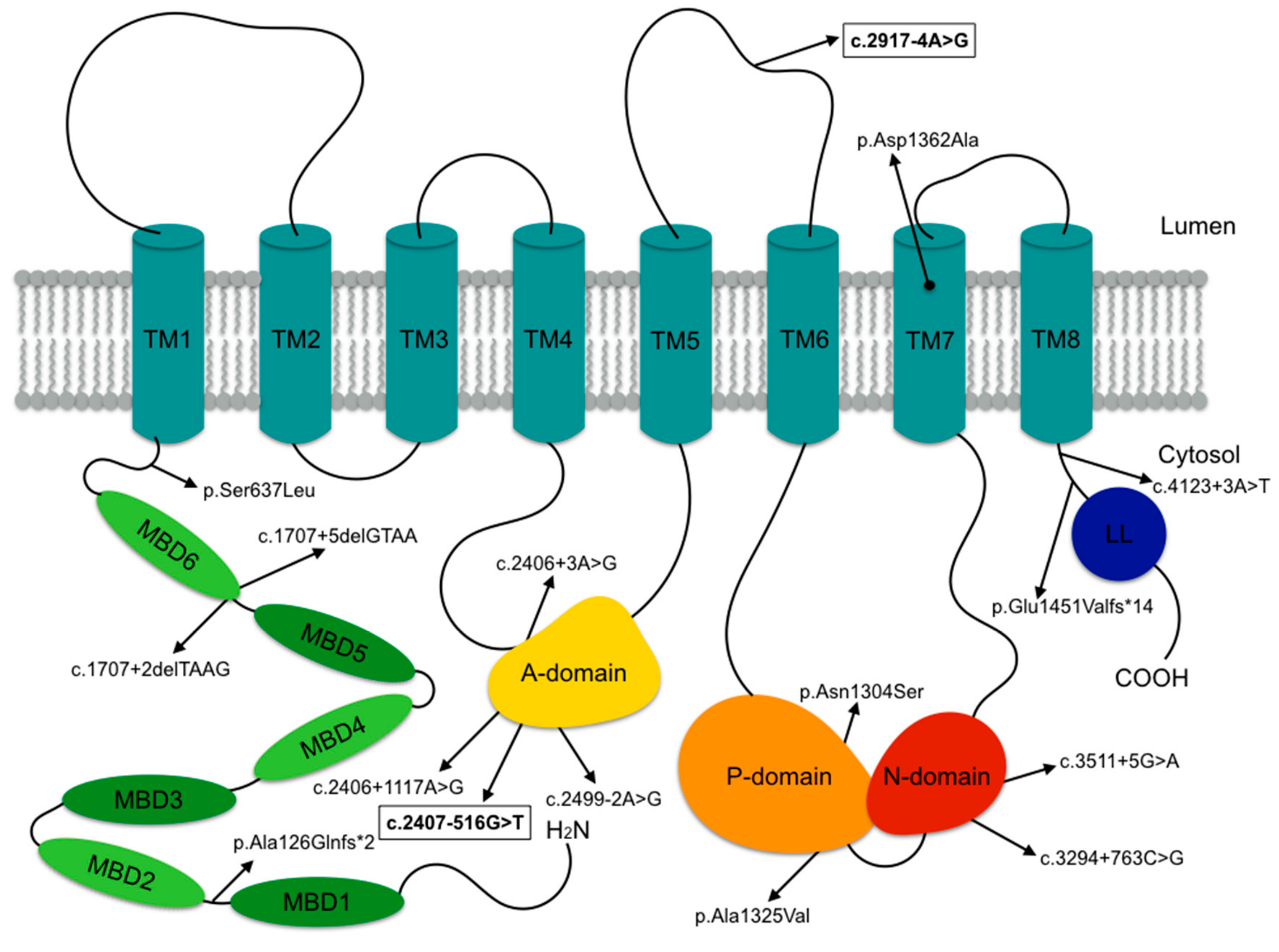
3.2. Molecular Characteristics
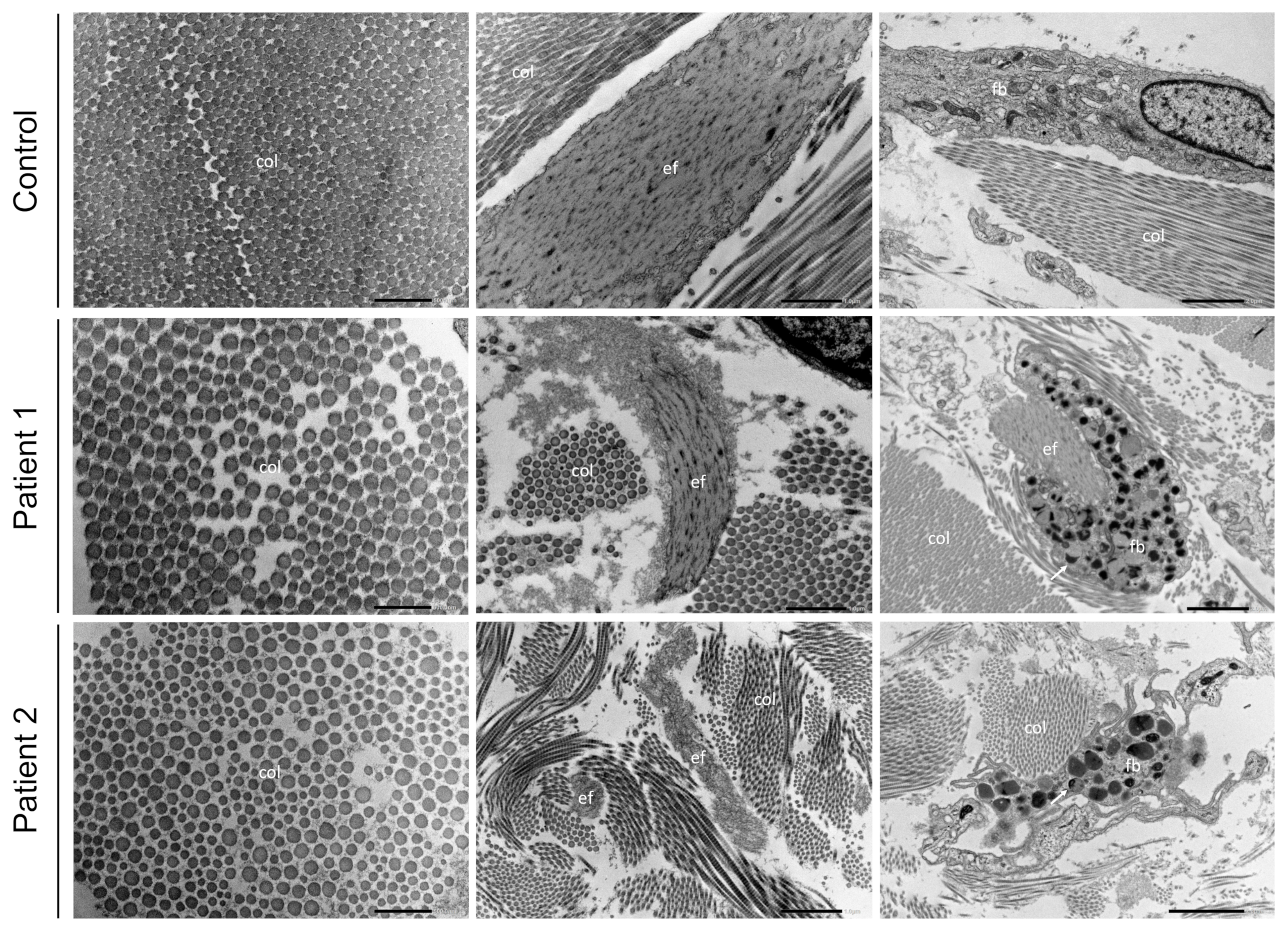
3.3. Transmission Electron Microscopy
3.4. Collagen Biochemistry
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tumer, Z.; Horn, N. Menkes disease: Recent advances and new aspects. J. Med. Genet. 1997, 344, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Tumer, Z.; Moller, L.B. Menkes disease. Eur. J. Hum. Genet. 2010, 185, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Kaler, S.G. ATP7A-related copper transport diseases-emerging concepts and future trends. Nat. Rev. Neurol. 2011, 71, 15–29. [Google Scholar] [CrossRef] [PubMed]
- Horn, N.; Tümer, Z. Menkes disease and the occipital horn syndrome. In Connective Tissue and Its Heritable Disorders; Royce, P.M., Steinman, B., Eds.; Wiley-Liss: New York, NY, USA, 2002; pp. 651–685. [Google Scholar]
- Sartoris, D.J.; Luzzatti, L.; Weaver, D.D.; Macfarlane, J.D.; Hollister, D.W.; Parker, B.R. Type IX Ehlers-Danlos syndrome. A new variant with pathognomonic radiographic features. Radiology 1984, 1523, 665–670. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Levinson, B.; Vulpe, C.; Whitney, S.; Gitschier, J.; Packman, S. Similar splicing mutations of the Menkes/mottled copper-transporting ATPase gene in occipital horn syndrome and the blotchy mouse. Am. J. Hum. Genet. 1995, 563, 570–576. [Google Scholar]
- Byers, P.H.; Siegel, R.C.; Holbrook, K.A.; Narayanan, A.S.; Bornstein, P.; Hall, J.G. X-linked cutis laxa: Defective cross-link formation in collagen due to decreased lysyl oxidase activity. N. Engl. J. Med. 1980, 3032, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Petris, M.J.; Strausak, D.; Mercer, J.F. The Menkes copper transporter is required for the activation of tyrosinase. Hum. Mol. Genet. 2000, 919, 2845–2851. [Google Scholar] [CrossRef] [PubMed]
- Kodama, H.; Okabe, I.; Yanagisawa, M.; Kodama, Y. Copper deficiency in the mitochondria of cultured skin fibroblasts from patients with Menkes syndrome. J. Inherit. Metab. Dis. 1989, 124, 386–389. [Google Scholar] [CrossRef]
- Royce, P.M.; Camakaris, J.; Danks, D.M. Reduced lysyl oxidase activity in skin fibroblasts from patients with Menkes’ syndrome. Biochem. J. 1980, 1922, 579–586. [Google Scholar] [CrossRef]
- Dagenais, S.L.; Adam, A.N.; Innis, J.W.; Glover, T.W. A novel frameshift mutation in exon 23 of ATP7A (MNK) results in occipital horn syndrome and not in Menkes disease. Am. J. Hum. Genet. 2001, 692, 420–427. [Google Scholar] [CrossRef]
- Kaler, S.G. Metabolic and molecular bases of Menkes disease and occipital horn syndrome. Pediatr. Dev. Pathol. 1998, 11, 85–98. [Google Scholar]
- Tumer, Z. An overview and update of ATP7A mutations leading to Menkes disease and occipital horn syndrome. Hum Mutat. 2013, 343, 417–429. [Google Scholar] [CrossRef] [PubMed]
- Hartwig, C.; Zlatic, S.A.; Wallin, M.; Vrailas-Mortimer, A.; Fahrni, C.J.; Faundez, V. Trafficking mechanisms of P-type ATPase copper transporters. Curr. Opin. Cell Biol. 2019, 59, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Polishchuk, R.; Lutsenko, S. Golgi in copper homeostasis: A view from the membrane trafficking field. Histochem. Cell Biol. 2013, 1403, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Dierick, H.A.; Ambrosini, L.; Spencer, J.; Glover, T.W.; Mercer, J.F.B. Molecular-Structure of the Menkes Disease Gene (ATP7A). Genomics 1995, 28, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Tumer, Z.; Vural, B.; Tonnesen, T.; Chelly, J.; Monaco, A.P.; Horn, N. Characterization of the exon structure of the Menkes disease gene using vectorette PCR. Genomics 1995, 263, 437–442. [Google Scholar] [CrossRef]
- Chelly, J.; Tumer, Z.; Tonnesen, T.; Petterson, A.; Ishikawa-Brush, Y.; Tommerup, N.; Horn, N.; Monaco, A.P. Isolation of a candidate gene for Menkes disease that encodes a potential heavy metal binding protein. Nat. Genet. 1993, 31, 14–19. [Google Scholar] [CrossRef]
- Mercer, J.F.B.; Livingston, J.; Hall, B.; Paynter, J.A.; Begy, C.; Chandrasekharappa, S.; Lockhart, P.; Grimes, A.; Bhave, M.; Siemieniak, D.; et al. Isolation of a Partial Candidate Gene for Menkes Disease by Positional Cloning. Nat. Genet. 1993, 31, 20–25. [Google Scholar] [CrossRef]
- Vulpe, C.; Levinson, B.; Whitney, S.; Packman, S.; Gitschier, J. Isolation of a candidate gene for Menkes disease and evidence that it encodes a copper-transporting ATPase. Nat. Genet. 1993, 31, 7–13. [Google Scholar] [CrossRef]
- Skjorringe, T.; Pedersen, P.A.; Thorborg, S.S.; Nissen, P.; Gourdon, P.; Moller, L.B. Characterization of ATP7A missense mutants suggests a correlation between intracellular trafficking and severity of Menkes disease. Sci. Rep.-Uk 2017, 7, 757. [Google Scholar] [CrossRef]
- De Bie, P.; Muller, P.; Wijmenga, C.; Klomp, L.W. Molecular pathogenesis of Wilson and Menkes disease: Correlation of mutations with molecular defects and disease phenotypes. J. Med. Genet. 2007, 4411, 673–688. [Google Scholar] [CrossRef] [PubMed]
- Gourdon, P.; Liu, X.Y.; Skjorringe, T.; Morth, J.P.; Moller, L.B.; Pedersen, B.P.; Nissen, P. Crystal structure of a copper-transporting PIB-type ATPase. Nature 2011, 475(7354), 59–64. [Google Scholar] [CrossRef] [PubMed]
- Strausak, D.; La Fontaine, S.; Hill, J.; Firth, S.D.; Lockhart, P.J.; Mercer, J.F.B. The role of GMXCXXC metal binding sites in the copper-induced redistribution of the Menkes protein. J. Biol. Chem. 1999, 27416, 11170–11177. [Google Scholar] [CrossRef] [PubMed]
- Gitschier, J.; Moffat, B.; Reilly, D.; Wood, W.I.; Fairbrother, W.J. Solution structure of the fourth metal-binding domain from the Menkes copper-transporting ATpase. Nat. Struct. Biol. 1998, 51, 47–54. [Google Scholar] [CrossRef]
- Das, S.; Levinson, B.; Whitney, S.; Vulpe, C.; Packman, S.; Gitschier, J. Diverse mutations in patients with Menkes disease often lead to exon skipping. Am. J. Hum. Genet. 1994, 555, 883–889. [Google Scholar]
- Kaler, S.G.; Gallo, L.K.; Proud, V.K.; Percy, A.K.; Mark, Y.; Segal, N.A.; Goldstein, D.S.; Holmes, C.S.; Gahl, W.A. Occipital horn syndrome and a mild Menkes phenotype associated with splice site mutations at the MNK locus. Nat. Genet. 1994, 82, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Ronce, N.; Moizard, M.P.; Robb, L.; Toutain, A.; Villard, L.; Moraine, C. A C2055T transition in exon 8 of the ATP7A gene is associated with exon skipping in an occipital horn syndrome family. Am. J. Hum. Genet. 1997, 611, 233–238. [Google Scholar] [CrossRef]
- Qi, M.; Byers, P.H. Constitutive skipping of alternatively spliced exon 10 in the ATP7A gene abolishes Golgi localization of the menkes protein and produces the occipital horn syndrome. Hum. Mol. Genet. 1998, 73, 465–469. [Google Scholar] [CrossRef]
- Borm, B.; Moller, L.B.; Hausser, I.; Emeis, M.; Baerlocher, K.; Horn, N.; Rossi, R. Variable clinical expression of an identical mutation in the ATP7A gene for Menkes disease/occipital horn syndrome in three affected males in a single family. J. Pediatr. 2004, 1451, 119–121. [Google Scholar] [CrossRef]
- Moller, L.B.; Tumer, Z.; Lund, C.; Petersen, C.; Cole, T.; Hanusch, R.; Seidel, J.; Jensen, L.R.; Horn, N. Similar splice-site mutations of the ATP7A gene lead to different phenotypes: Classical Menkes disease or occipital horn syndrome. Am. J. Hum. Genet. 2000, 664, 1211–1220. [Google Scholar] [CrossRef]
- Siegel, R.C. Lysyl oxidase. Int. Rev. Connect. Tissue Res. 1979, 8, 73–118. [Google Scholar] [PubMed]
- Biaggioni, I.; Goldstein, D.S.; Atkinson, T.; Robertson, D. Dopamine-beta-hydroxylase deficiency in humans. Neurology 1990, 402, 370–373. [Google Scholar] [CrossRef] [PubMed]
- Robertson, D.; Goldberg, M.R.; Onrot, J.; Hollister, A.S.; Wiley, R.; Thompson, J.G., Jr.; Robertson, R.M. Isolated failure of autonomic noradrenergic neurotransmission. Evidence for impaired beta-hydroxylation of dopamine. N. Engl. J. Med. 1986, 31423, 1494–1497. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.H.; Kodama, H.; Murata, Y.; Mochizuki, D.; Yanagawa, Y.; Ushijima, H.; Shiba, T.; Lee, C.C. ATP7A gene mutations in 16 patients with Menkes disease and a patient with occipital horn syndrome. Am. J. Med. Genet. 2001, 993, 217–222. [Google Scholar] [CrossRef]
- Wakai, S.; Ishikawa, Y.; Nagaoka, M.; Okabe, M.; Minami, R.; Hayakawa, T. Central nervous system involvement and generalized muscular atrophy in occipital horn syndrome: Ehlers-Danlos type IX. A first Japanese case. J. Neurol. Sci. 1993, 1161, 1–5. [Google Scholar] [CrossRef]
- Proud, V.K.; Mussell, H.G.; Kaler, S.G.; Young, D.W.; Percy, A.K. Distinctive Menkes disease variant with occipital horns: Delineation of natural history and clinical phenotype. Am. J. Med. Genet. 1996, 651, 44–51. [Google Scholar] [CrossRef]
- Levinson, B.; Conant, R.; Schnur, R.; Das, S.; Packman, S.; Gitschier, J. A repeated element in the regulatory region of the MNK gene and its deletion in a patient with occipital horn syndrome. Hum. Mol. Genet. 1996, 511, 1737–1742. [Google Scholar] [CrossRef]
- De Paepe, A.; Loeys, B.; Devriendt, K.; Fryns, J.P. Occipital Horn syndrome in a 2-year-old boy. Clin. Dysmorphol. 1999, 83, 179–183. [Google Scholar] [CrossRef]
- Yasmeen, S.; Lund, K.; De Paepe, A.; De Bie, S.; Heiberg, A.; Silva, J.; Martins, M.; Skjorringe, T.; Moller, L.B. Occipital horn syndrome and classical Menkes Syndrome caused by deep intronic mutations, leading to the activation of ATP7A pseudo-exon. Eur. J. Hum. Genet (EJHG) 2014, 224, 517–521. [Google Scholar] [CrossRef]
- Tang, J.; Robertson, S.; Lem, K.E.; Godwin, S.C.; Kaler, S.G. Functional copper transport explains neurologic sparing in occipital horn syndrome. Genet. Med. 2006, 811, 711–718. [Google Scholar] [CrossRef]
- De Gemmis, P.; Enzo, M.V.; Lorenzetto, E.; Cattelan, P.; Segat, D.; Hladnik, U. 13 novel putative mutations in ATP7A found in a cohort of 25 Italian families. Metab. Brain Dis. 2017, 324, 1173–1183. [Google Scholar] [CrossRef] [PubMed]
- Donsante, A.; Tang, J.; Godwin, S.C.; Holmes, C.S.; Goldstein, D.S.; Bassuk, A.; Kaler, S.G. Differences in ATP7A gene expression underlie intrafamilial variability in Menkes disease/occipital horn syndrome. J. Med. Genet. 2007, 448, 492–497. [Google Scholar] [CrossRef] [PubMed]
- Bazzocchi, A.; Femia, R.; Feraco, P.; Battista, G.; Canini, R.; Guglielmi, G. Occipital horn syndrome in a woman: Skeletal radiological findings. Skeletal Radiol. 2011, 4011, 1491–1494. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, E.; Kodama, H. Effects of disulfiram treatment in patients with Menkes disease and occipital horn syndrome. J. Trace Elements Med. Biol. 2012, 26, 102–104. [Google Scholar] [CrossRef] [PubMed]
- Nascimento, A.; Rego Sousa, P.; Ortez, C.; Boronat, S.; Rebello, M.; Jimenez-Mallabrera, C.; Jou, C.; Rovira, C.; Colomer, J. Expanding the phenotype of ATP7A-related copper transport diseases: Response to copper treatment. J. Inherit Metab. Dis. 2013, 36 (Suppl. 2), S91–S342. [Google Scholar]
- Legros, L.; Revencu, N.; Nassogne, M.C.; Wese, F.X.; Feyaerts, A. Multiple bladder diverticula caused by occipital horn syndrome. Arch. Pediatr. 2015, 2211, 1147–1150. [Google Scholar] [CrossRef] [PubMed]
- Louro, P.; Ramos, L.; Oliveira, R.; Henriques, M.; Pereira, E.; Kaler, S.G.; Pereira, J.; Diogo, L.; Garcia, P. Inherited Disorders of Metal Metabolism: A Rare Case of Occipital Horn Syndrome. In Proceedings of the 13th International Congress of Inborn Errors of Metabolism, Rio De Janeiro, Brazil, 5–8 September 2017. [Google Scholar]
- Palcic, I.; Cvitkovic Roic, A.; Roic, G.; Krakar, G.; Jaklin Kekez, A. (Eds.) Bladder diverticula caused by occipital horn syndrome—A case report. In Proceedings of the 50th Anniversary meeting of the European Society of Paediatric Nephrology, Glasglow, UK, 6–7 September 2017. [Google Scholar]
- Htut, E.P.; Offiah, A.C.; Holden, S.; Shenker, N.G. Compelling case of copper conundrum; Occipital horn syndrome. Rheumatology 2017, 56 (Suppl. 2). [Google Scholar] [CrossRef]
- Martin-Santiago, A.; Escudero-Gongora, M.M.; Giacaman, A.; Bauza, A.; Saus, C.; Roldan, J.; Rosell, J. Cutis laxa and copper transport anomalies, one gene, two ends of the spectrum. Pediatr. Dermatol. 2017, 34, S25. [Google Scholar]
- Nuytinck, L.; Narcisi, P.; Nicholls, A.; Renard, J.P.; Pope, F.M.; De Paepe, A. Detection and characterisation of an overmodified type III collagen by analysis of non-cutaneous connective tissues in a patient with Ehlers-Danlos syndrome IV. J. Med. Genet. 1992, 296, 375–380. [Google Scholar] [CrossRef]
- Syx, D.; De Wandele, I.; Symoens, S.; De Rycke, R.; Hougrand, O.; Voermans, N.; De Paepe, A.; Malfait, F. Bi-allelic AEBP1 mutations in two patients with Ehlers-Danlos syndrome. Hum. Mol. Genet. 2019, 2811, 1853–1864. [Google Scholar] [CrossRef]
- Tsukahara, M.; Imaizumi, K.; Kawai, S.; Kajii, T. Occipital horn syndrome: Report of a patient and review of the literature. Clin. Genet. 1994, 451, 32–35. [Google Scholar] [CrossRef]
- Lazoff, S.G.; Rybak, J.J.; Parker, B.R.; Luzzatti, L. Skeletal dysplasia, occipital horns, diarrhea and obstructive uropathy—A new hereditary syndrome. Birth Defects Orig. Article Series. 1975, 115, 71–74. [Google Scholar]
- Callewaert, B.; Su, C.T.; Van Damme, T.; Vlummens, P.; Malfait, F.; Vanakker, O.; Schulz, B.; Mac Neal, M.; Davis, E.C.; Lee, J.G.; et al. Comprehensive clinical and molecular analysis of 12 families with type 1 recessive cutis laxa. Hum Mutat. 2013, 341, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Malfait, F.; De Coster, P.; Hausser, I.; van Essen, A.J.; Franck, P.; Colige, A.; Nusgens, B.; Martens, L.; De Paepe, A. The natural history, including orofacial features of three patients with Ehlers-Danlos syndrome, dermatosparaxis type (EDS type VIIC). Am. J. Med. Genet. Part A. 2004, 1311, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Wuyts, W.; Van Hul, W.; De Boulle, K.; Hendrickx, J.; Bakker, E.; Vanhoenacker, F.; Mollica, F.; Lüdecke, H.J.; Sayli, B.S.; Pazzaglia, U.E.; et al. Mutations in the EXT1 and EXT2 genes in hereditary multiple exostoses. Am. J. Hum. Genet. 1998, 622, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Jurik, A.G.; Jorgensen, P.H.; Mortensen, M.M. Whole-body MRI in assessing malignant transformation in multiple hereditary exostoses and enchondromatosis: Audit results and literature review. Skeletal Radiol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Moller, L.B. Small amounts of functional ATP7A protein permit mild phenotype. J. Trace Elem. Med. Biol. 2015, 31, 173–177. [Google Scholar] [CrossRef]
- Moller, J.V.; Juul, B.; le Maire, M. Structural organization, ion transport, and energy transduction of P-type ATPases. Biochim. Biophys. Acta 1996, 12861, 1–51. [Google Scholar] [CrossRef]
- Paulsen, M.; Lund, C.; Akram, Z.; Winther, J.R.; Horn, N.; Moller, L.B. Evidence that translation reinitiation leads to a partially functional Menkes protein containing two copper-binding sites. Am. J. Hum. Genet. 2006, 792, 214–229. [Google Scholar] [CrossRef]
- Uitto, J.; Li, Q.; Urban, Z. The complexity of elastic fibre biogenesis in the skin--A perspective to the clinical heterogeneity of cutis laxa. Exp. Dermatol. 2013, 222, 88–92. [Google Scholar] [CrossRef]
- Symoens, S.; Renard, M.; Bonod-Bidaud, C.; Syx, D.; Vaganay, E.; Malfait, F.; Ricard-Blum, S.; Kessler, E.; Van Laer, L.; Coucke, P.; et al. Identification of binding partners interacting with the alpha1-N-propeptide of type V collagen. Biochem. J. 2011, 4332, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Vogel, A.; Holbrook, K.A.; Steinmann, B.; Gitzelmann, R.; Byers, P.H. Abnormal collagen fibril structure in the gravis form (type I) of Ehlers-Danlos syndrome. Laboratory investigation. J. Techn. Methods Pathol. 1979, 402, 201–206. [Google Scholar]
- Malfait, F.; Coucke, P.; Symoens, S.; Loeys, B.; Nuytinck, L.; De Paepe, A. The molecular basis of classic Ehlers-Danlos syndrome: A comprehensive study of biochemical and molecular findings in 48 unrelated patients. Hum. Mutat. 2005, 251, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Birk, D.E. Type V collagen: Heterotypic type I/V collagen interactions in the regulation of fibril assembly. Micron (Oxford, England: 1993) 2001, 323, 223–237. [Google Scholar] [CrossRef]
- Hausser, I.; Anton-Lamprecht, I. Differential ultrastructural aberrations of collagen fibrils in Ehlers-Danlos syndrome types I-IV as a means of diagnostics and classification. Hum. Genet. 1994, 934, 394–407. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Total (Percentage) | |
|---|---|
| Craniofacial | |
| Long face | 6/13 (46%) |
| Large ears | 5/13 (38%) |
| Sagging/ full cheeks | 5/11 (45%) |
| Hair abnormalities | 14/19 (74%) |
| Pili torti on trichoscopy | 7/10 (70%) |
| Connective tissue | |
| Cutis laxa | 25/27 (93%) |
| Inguinal hernia | 15/24 (63%) |
| Umbilical hernia | 1/22 (5%) |
| Osteoarticular | |
| Occipital horns | 25/26 (96%) |
| Radial/tibial exostoses | 6/16 (38%) |
| Hammer-shaped clavicula | 9/19 (47%) |
| Bowing of long bones | 4/17 (23%) |
| Mid-diaphyseal broadening | 2/17 (11%) |
| Rounding of the iliac wings | 3/17 (18%) |
| Coxa valga | 6/16 (38%) |
| Genu valgum | 6/17 (35%) |
| Metaphyseal spurring | 2/16 (13%) |
| Scoliosis | 8/21 (38%) |
| Pectus deformity | 13/23 (57%) |
| Dislocations | 12/20 (60%) |
| Contractures of large joints | 3/19 (16%) |
| Joint hyperlaxity | 16/26 (62%) |
| Fractures | 2/21 (10%) |
| Neurological | |
| Intellectual disability | 17/33 (52%) |
| Seizures | 5/25 (20%) |
| Muscle hypotonia | 10/25 (40%) |
| Stroke | 1/23 (4%) |
| Cardiovascular | |
| Aneurysm formation | 5/7 (71%) |
| Dilatation of the large veins | 2/6 (33%) |
| Intracranial tortuosity | 7/11 (64%) |
| Extracranial tortuosity | 3/4 (75%) |
| Dysautonomia | 13/15 (87%) |
| Urogenital | |
| Bladder diverticula | 25/30 (83%) |
| Renal abnormalities | 6/20 (30%) |
| Urinary tract infections | 17/23 (74%) |
| Vesicourethral reflux | 7/23 (30%) |
| Laboratory findings | |
| Serum copper | 20/28 (71%) |
| Serum ceruloplasmin | 20/27 (74%) |
| Initial Presentation | Number of Patients |
|---|---|
| Neurological | 11 |
| Seizures | 1 |
| Developmental delay | 4 |
| Hypotonia | 6 |
| Connective tissue | 9 |
| Cephalhematoma | 4 |
| Generalized CTD | 3 |
| Inguinal hernia | 2 |
| Urogenital | 5 |
| Bladder diverticula | 3 |
| Urinary infections | 2 |
| Skeletal | 3 |
| Pectus deformity | 1 |
| Skeletal dysplasia | 1 |
| Joint pain | 1 |
| Other | 3 |
| Vomiting and diarrhea | 1 |
| Dysautonomia | 1 |
| Apnea | 1 |
| Segregation analysis | 1 |
| Unknown | 2 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beyens, A.; Van Meensel, K.; Pottie, L.; De Rycke, R.; De Bruyne, M.; Baeke, F.; Hoebeke, P.; Plasschaert, F.; Loeys, B.; De Schepper, S.; et al. Defining the Clinical, Molecular and Ultrastructural Characteristics in Occipital Horn Syndrome: Two New Cases and Review of the Literature. Genes 2019, 10, 528. https://doi.org/10.3390/genes10070528
Beyens A, Van Meensel K, Pottie L, De Rycke R, De Bruyne M, Baeke F, Hoebeke P, Plasschaert F, Loeys B, De Schepper S, et al. Defining the Clinical, Molecular and Ultrastructural Characteristics in Occipital Horn Syndrome: Two New Cases and Review of the Literature. Genes. 2019; 10(7):528. https://doi.org/10.3390/genes10070528
Chicago/Turabian StyleBeyens, Aude, Kyaran Van Meensel, Lore Pottie, Riet De Rycke, Michiel De Bruyne, Femke Baeke, Piet Hoebeke, Frank Plasschaert, Bart Loeys, Sofie De Schepper, and et al. 2019. "Defining the Clinical, Molecular and Ultrastructural Characteristics in Occipital Horn Syndrome: Two New Cases and Review of the Literature" Genes 10, no. 7: 528. https://doi.org/10.3390/genes10070528
APA StyleBeyens, A., Van Meensel, K., Pottie, L., De Rycke, R., De Bruyne, M., Baeke, F., Hoebeke, P., Plasschaert, F., Loeys, B., De Schepper, S., Symoens, S., & Callewaert, B. (2019). Defining the Clinical, Molecular and Ultrastructural Characteristics in Occipital Horn Syndrome: Two New Cases and Review of the Literature. Genes, 10(7), 528. https://doi.org/10.3390/genes10070528