Identification of Main Genetic Causes Responsible for Non-Syndromic Hearing Loss in a Peruvian Population


 ,
,
Abstract
:1. Introduction
2. Methods
2.1. Subjects
2.2. Sanger Sequencing of GJB2 Exon 1 and Exon 2
2.3. Whole Genome Sequencing Analysis
2.4. Genome-Wide Genotyping and Quality Control
2.5. Admixture Analysis
2.6. Local Ancestry Analysis
3. Results
3.1. GJB2 Sequencing
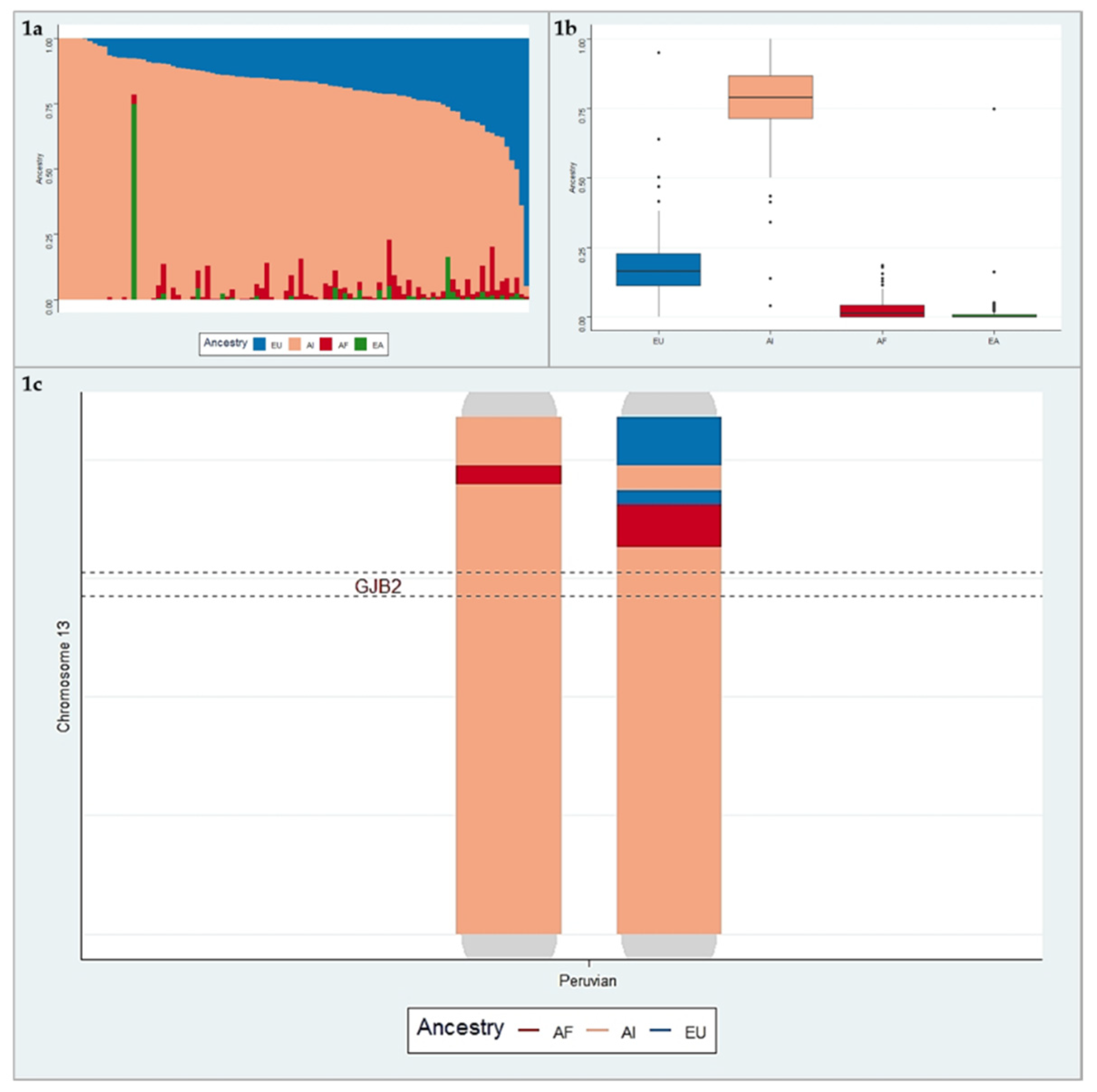
3.2. Ancestry Analysis
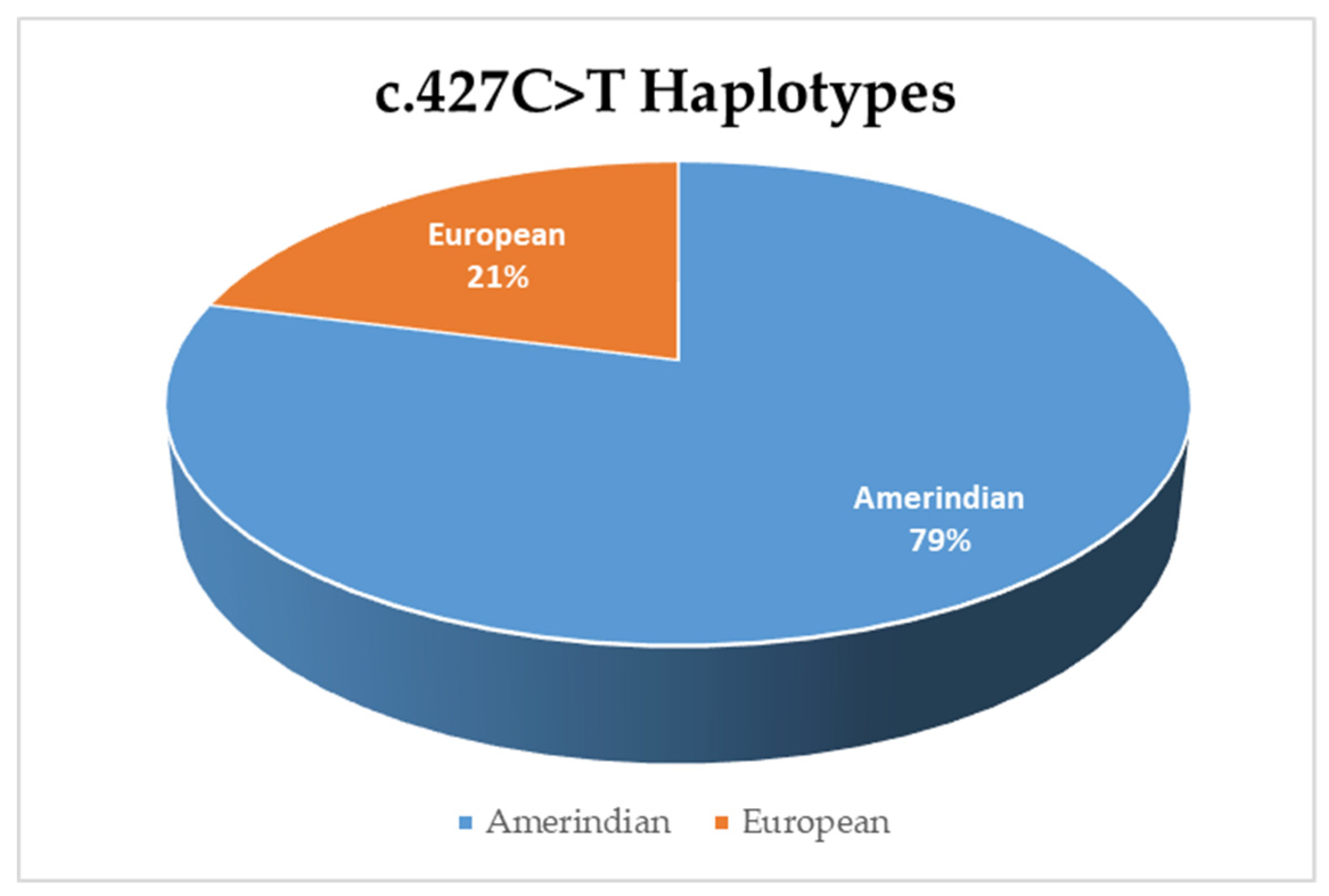
3.3. Haplotype Analysis for Variant c.427C>T
3.4. Whole Genome Sequencing
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Deafness and hearing loss. Available online: http://www.who.int/news-room/fact-sheets/detail/deafness-and-hearing-loss (accessed on 30 October 2018).
- Shearer, A.E.; Hildebrand, M.S.; Smith, R.J. Hereditary Hearing Loss and Deafness Overview. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Egilmez, O.K.; Kalcioglu, M.T. Genetics of Nonsyndromic Congenital Hearing Loss. Available online: https://www.hindawi.com/journals/scientifica/2016/7576064/ (accessed on 30 October2018).
- Welcome to the Hereditary Hearing Loss Homepage | Hereditary Hearing Loss Homepage. Available online: https://hereditaryhearingloss.org/ (accessed on 7 April 2019).
- Yan, D.; Tekin, D.; Bademci, G.; Foster, J.; Cengiz, F.B.; Kannan-Sundhari, A.; Guo, S.; Mittal, R.; Zou, B.; Grati, M.; et al. Spectrum of DNA variants for non-syndromic deafness in a large cohort from multiple continents. Hum. Genet. 2016, 135, 953–961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, R.J.; Jones, M.-K.N. Nonsyndromic Hearing Loss and Deafness, DFNB1. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Del Castillo, F.J.; Del Castillo, I. DFNB1 Non-syndromic Hearing Impairment: Diversity of Mutations and Associated Phenotypes. Front. Mol. Neurosci. 2017, 10, 428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snoeckx, R.L.; Huygen, P.L.M.; Feldmann, D.; Marlin, S.; Denoyelle, F.; Waligora, J.; Mueller-Malesinska, M.; Pollak, A.; Ploski, R.; Murgia, A.; et al. GJB2 Mutations and Degree of Hearing Loss: A Multicenter Study. Am. J. Hum. Genet. 2005, 77, 945–957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohtsuka, A.; Yuge, I.; Kimura, S.; Namba, A.; Abe, S.; Van Laer, L.; Van Camp, G.; Usami, S. GJB2 deafness gene shows a specific spectrum of mutations in Japan, including a frequent founder mutation. Hum. Genet. 2003, 112, 329–333. [Google Scholar] [PubMed]
- Taniguchi, M.; Matsuo, H.; Shimizu, S.; Nakayama, A.; Suzuki, K.; Hamajima, N.; Shinomiya, N.; Nishio, S.; Kosugi, S.; Usami, S.-I.; et al. Carrier frequency of the GJB2 mutations that cause hereditary hearing loss in the Japanese population. J. Hum. Genet. 2015, 60, 613–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, J.; Ying, Z.; Cai, Z.; Sun, D.; He, Z.; Gao, Y.; Zhang, T.; Zhu, Y.; Chen, Y.; Guan, M.-X. GJB2 Mutation Spectrum and Genotype-Phenotype Correlation in 1067 Han Chinese Subjects with Non-Syndromic Hearing Loss. PloS One 2015, 10, e0128691. [Google Scholar] [CrossRef]
- Kim, S.Y.; Kim, A.R.; Han, K.H.; Kim, M.Y.; Jeon, E.-H.; Koo, J.-W.; Oh, S.H.; Choi, B.Y. Residual Hearing in DFNB1 Deafness and Its Clinical Implication in a Korean Population. PloS One 2015, 10, e0125416. [Google Scholar] [CrossRef]
- de la Luz Arenas-Sordo, M.; Menendez, I.; Hernández-Zamora, E.; Sirmaci, A.; Gutiérrez-Tinajero, D.; McGetrick, M.; Murphy-Ruiz, P.; Leyva-Juárez, X.; Huesca-Hernández, F.; Dominguez-Aburto, J.; et al. Unique spectrum of GJB2 mutations in Mexico. Int. J. Pediatr. Otorhinolaryngol. 2012, 76, 1678–1680. [Google Scholar] [CrossRef]
- Gravina, L.P.; Foncuberta, M.E.; Prieto, M.E.; Garrido, J.; Barreiro, C.; Chertkoff, L. Prevalence of DFNB1 mutations in Argentinean children with non-syndromic deafness. Report of a novel mutation in GJB2. Int. J. Pediatr. Otorhinolaryngol. 2010, 74, 250–254. [Google Scholar] [CrossRef]
- Instituto Nacional de Estadística e Informática. Primera Encuesta Nacional Especializada sobre DISCAPACIDAD 2012; Instituto Nacional de Estadística e Informática: Lima, Peru, 2014. [Google Scholar]
- Harris, D.N.; Song, W.; Shetty, A.C.; Levano, K.S.; Cáceres, O.; Padilla, C.; Borda, V.; Tarazona, D.; Trujillo, O.; Sanchez, C.; et al. Evolutionary genomic dynamics of Peruvians before, during, and after the Inca Empire. Proc. Natl. Acad. Sci. USA 2018, 115, E6526–E6535. [Google Scholar] [CrossRef] [Green Version]
- Sandoval, J.R.; Salazar-Granara, A.; Acosta, O.; Castillo-Herrera, W.; Fujita, R.; Pena, S.D.J.; Santos, F.R. Tracing the genomic ancestry of Peruvians reveals a major legacy of pre-Columbian ancestors. J. Hum. Genet. 2013, 58, 627–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chacón Villanueva, R.; Badillo, R.; Cornejo-Olivas, M.; Ortega, O.; Marca, V.; Descailleaux, R.; Rubio, J.; Mazzetti-Soler, P. Mutations of Connexin Genes 26(GJB2) and 30(GJB6) in a Peruvian Deaf Population. In Proceedings of the 12th International Congress of Human Genetics and the American Society of Human Genetics 61st Annual Meeting, Montreal, QC, Canada, 11–15 October 2011. [Google Scholar]
- Tekin, M.; Xia, X.-J.; Erdenetungalag, R.; Cengiz, F.B.; White, T.W.; Radnaabazar, J.; Dangaasuren, B.; Tastan, H.; Nance, W.E.; Pandya, A. GJB2 mutations in Mongolia: complex alleles, low frequency, and reduced fitness of the deaf. Ann. Hum. Genet. 2010, 74, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. Off. J. Am. Coll. Med. Genet. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Liang, X.; Xuan, Y.; Geng, C.; Li, Y.; Lu, H.; Qu, S.; Mei, X.; Chen, H.; Yu, T.; et al. A reference human genome dataset of the BGISEQ-500 sequencer. GigaScience 2017, 6, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinforma. Oxf. Engl. 2010, 26, 589–595. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Abyzov, A.; Urban, A.E.; Snyder, M.; Gerstein, M. CNVnator: an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res. 2011, 21, 974–984. [Google Scholar] [CrossRef]
- Chen, K.; Wallis, J.W.; McLellan, M.D.; Larson, D.E.; Kalicki, J.M.; Pohl, C.S.; McGrath, S.D.; Wendl, M.C.; Zhang, Q.; Locke, D.P.; et al. BreakDancer: an algorithm for high-resolution mapping of genomic structural variation. Nat. Methods 2009, 6, 677–681. [Google Scholar] [CrossRef] [Green Version]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [Green Version]
- Reich, D.; Patterson, N.; Campbell, D.; Tandon, A.; Mazieres, S.; Ray, N.; Parra, M.V.; Rojas, W.; Duque, C.; Mesa, N.; et al. Reconstructing Native American population history. Nature 2012, 488, 370–374. [Google Scholar] [CrossRef] [PubMed]
- Delaneau, O.; Marchini, J. 1000 Genomes Project Consortium; 1000 Genomes Project Consortium Integrating sequence and array data to create an improved 1000 Genomes Project haplotype reference panel. Nat. Commun. 2014, 5, 3934. [Google Scholar] [CrossRef] [PubMed]
- Maples, B.K.; Gravel, S.; Kenny, E.E.; Bustamante, C.D. RFMix: a discriminative modeling approach for rapid and robust local-ancestry inference. Am. J. Hum. Genet. 2013, 93, 278–288. [Google Scholar] [CrossRef] [PubMed]
- Rajabli, F.; Feliciano, B.E.; Celis, K.; Hamilton-Nelson, K.L.; Whitehead, P.L.; Adams, L.D.; Bussies, P.L.; Manrique, C.P.; Rodriguez, A.; Rodriguez, V.; et al. Ancestral origin of ApoE ε4 Alzheimer disease risk in Puerto Rican and African American populations. PLoS Genet. 2018, 14, e1007791. [Google Scholar] [CrossRef] [PubMed]
- Hamelmann, C.; Amedofu, G.K.; Albrecht, K.; Muntau, B.; Gelhaus, A.; Brobby, G.W.; Horstmann, R.D. Pattern of connexin 26 (GJB2) mutations causing sensorineural hearing impairment in Ghana. Hum. Mutat. 2001, 18, 84–85. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, C.; Funayama, M.; Li, Y.; Kamiya, K.; Kawano, A.; Suzuki, M.; Hattori, N.; Ikeda, K. Prevalence of GJB2 causing recessive profound non-syndromic deafness in Japanese children. Int. J. Pediatr. Otorhinolaryngol. 2011, 75, 211–214. [Google Scholar] [CrossRef] [PubMed]
- Paz-y-Miño, C.; Beaty, D.; López-Cortés, A.; Proaño, I. Frequency of GJB2 and del(GJB6-D13S1830) mutations among an Ecuadorian mestizo population. Int. J. Pediatr. Otorhinolaryngol. 2014, 78, 1648–1654. [Google Scholar] [CrossRef]
- Hernández-Juárez, A.A.; Lugo-Trampe, J.J.; Campos-Acevedo, L.D.; Lugo-Trampe, A.; Treviño-González, J.L.; de-la-Cruz-Ávila, I.; Martínez-de-Villarreal, L.E. GJB2 and GJB6 mutations are an infrequent cause of autosomal-recessive nonsyndromic hearing loss in residents of Mexico. Int. J. Pediatr. Otorhinolaryngol. 2014, 78, 2107–2112. [Google Scholar]
- Loeza-Becerra, F.; Rivera-Vega, M.R.; Martínez-Saucedo, M.; Gonzalez-Huerta, L.M.; Urueta-Cuellar, H.; Berrruecos-Villalobos, P.; Cuevas-Covarrubias, S. Particular distribution of the GJB2/GJB6 gene mutations in Mexican population with hearing impairment. Int. J. Pediatr. Otorhinolaryngol. 2014, 78, 1057–1060. [Google Scholar] [CrossRef]
- de Oliveira, C.A.; Alexandrino, F.; Christiani, T.V.; Steiner, C.E.; Cunha, J.L.R.; Guerra, A.T.M.; Sartorato, E.L. Molecular genetics study of deafness in Brazil: 8-year experience. Am. J. Med. Genet. A. 2007, 143A, 1574–1579. [Google Scholar] [CrossRef]
- Utrera, R.; Ridaura, V.; Rodríguez, Y.; Rojas, M.J.; Mago, L.; Angeli, S.; Henríquez, O. Detection of the 35delG/GJB2 and del(GJB6-D13S1830) mutations in Venezuelan patients with autosomal recessive nonsyndromic hearing loss. Genet. Test. 2007, 11, 347–352. [Google Scholar] [CrossRef] [PubMed]
- Tamayo, M.L.; Olarte, M.; Gelvez, N.; Gómez, M.; Frías, J.L.; Bernal, J.E.; Florez, S.; Medina, D. Molecular studies in the GJB2 gene (Cx26) among a deaf population from Bogotá, Colombia: results of a screening program. Int. J. Pediatr. Otorhinolaryngol. 2009, 73, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Angeli, S.I. Phenotype/genotype correlations in a DFNB1 cohort with ethnical diversity. Laryngoscope 2008, 118, 2014–2023. [Google Scholar] [CrossRef] [PubMed]
- King, P.J.; Ouyang, X.; Du, L.; Yan, D.; Angeli, S.I.; Liu, X.Z. Etiologic Diagnosis of Nonsyndromic Genetic Hearing Loss in Adult vs Pediatric Populations. Otolaryngol.--Head Neck Surg. Off. J. Am. Acad. Otolaryngol.-Head Neck Surg. 2012, 147, 932–936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rehman, A.U.; Bird, J.E.; Faridi, R.; Shahzad, M.; Shah, S.; Lee, K.; Khan, S.N.; Imtiaz, A.; Ahmed, Z.M.; Riazuddin, S.; et al. Mutational Spectrum of MYO15A and the Molecular Mechanisms of DFNB3 Human Deafness. Hum. Mutat. 2016, 37, 991–1003. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Guan, J.; Wang, H.; Yin, L.; Wang, D.; Zhao, L.; Zhou, H.; Wang, Q. Genotype-phenotype correlation analysis of MYO15A variants in autosomal recessive non-syndromic hearing loss. BMC Med. Genet. 2019, 20, 60. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Pathogenic Variant | Heterozygous | Homozygous | Total of Families | Frequency * |
|---|---|---|---|---|
| c.427C>T; p.(Arg143Trp) | 19 | 1 | 20 | 22.6% |
| c.35delG; p.(Gly12fs) | 10 | 3 | 13 | 17.2% |
| c.94C>A; p.(Arg32Ser) | 9 | 1 | 10 | 11.8% |
| c.59T>C; p.(Ile20Thr) | 8 | 1 | 9 | 10.8% |
| c.35G>T; p.(Gly12Val) | 3 | 3 | 6 | 9.7% |
| c.283G>A; p.(Val95Met) | 6 | 0 | 6 | 6.5% |
| c.645delT; p.(Arg216fs) | 6 | 0 | 6 | 6.5% |
| c.19C>T; p.(Gln7Ter) | 5 | 0 | 5 | 5.4% |
| c.167delT; p.(Leu56fs) | 3 | 0 | 3 | 3.2% |
| c.109G>A; p.(Val37Ile) | 2 | 0 | 2 | 2.2% |
| c.29T>C; p.(Leu10Pro) | 1 | 0 | 1 | 1.1% |
| c.235delC; p.(Leu79fs) | 1 | 0 | 1 | 1.1% |
| c.617A>G; p.(Asn206Ser) | 1 | 0 | 1 | 1.1% |
| c.232dupG; p.(Ala78fs) | 1 | 0 | 1 | 1.1% |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Figueroa-Ildefonso, E.; Bademci, G.; Rajabli, F.; Cornejo-Olivas, M.; Chacón Villanueva, R.D.; Badillo-Carrillo, R.; Inca-Martinez, M.; Milla Neyra, K.; Sineni, C.; Tekin, M. Identification of Main Genetic Causes Responsible for Non-Syndromic Hearing Loss in a Peruvian Population. Genes 2019, 10, 581. https://doi.org/10.3390/genes10080581
Figueroa-Ildefonso E, Bademci G, Rajabli F, Cornejo-Olivas M, Chacón Villanueva RD, Badillo-Carrillo R, Inca-Martinez M, Milla Neyra K, Sineni C, Tekin M. Identification of Main Genetic Causes Responsible for Non-Syndromic Hearing Loss in a Peruvian Population. Genes. 2019; 10(8):581. https://doi.org/10.3390/genes10080581
Chicago/Turabian StyleFigueroa-Ildefonso, Erick, Guney Bademci, Farid Rajabli, Mario Cornejo-Olivas, Ruy Diego Chacón Villanueva, Rodolfo Badillo-Carrillo, Miguel Inca-Martinez, Karina Milla Neyra, Claire Sineni, and Mustafa Tekin. 2019. "Identification of Main Genetic Causes Responsible for Non-Syndromic Hearing Loss in a Peruvian Population" Genes 10, no. 8: 581. https://doi.org/10.3390/genes10080581
APA StyleFigueroa-Ildefonso, E., Bademci, G., Rajabli, F., Cornejo-Olivas, M., Chacón Villanueva, R. D., Badillo-Carrillo, R., Inca-Martinez, M., Milla Neyra, K., Sineni, C., & Tekin, M. (2019). Identification of Main Genetic Causes Responsible for Non-Syndromic Hearing Loss in a Peruvian Population. Genes, 10(8), 581. https://doi.org/10.3390/genes10080581