The Genomics of Streptococcus pneumoniae Carriage Isolates from UK Children and Their Household Contacts, Pre-PCV7 to Post-PCV13

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
3. Results
3.1. Serotyping
3.1.1. Population Structure (MLST and PopPUNK)
3.1.2. PopPUNK Analysis and Comparison with Global Datasets
3.2. Pangenome Analysis
3.3. Antibiotic Resistance Markers
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Miller, E.; Andrews, N.J.; Waight, P.A.; Slack, M.P.; George, R.C. Herd immunity and serotype replacement 4 years after seven-valent pneumococcal conjugate vaccination in England and Wales: An observational cohort study. Lancet Infect. Dis. 2011, 11, 760–768. [Google Scholar] [CrossRef]
- Hussain, M.; Melegaro, A.; Pebody, R.G.; George, R.; Edmunds, W.J.; Talukdar, R.; Martin, S.A.; Efstratiou, A.; Miller, E. A longitudinal household study of Streptococcus pneumoniae nasopharyngeal carriage in a UK setting. Epidemiol. Infect. 2005, 133, 891–898. [Google Scholar] [CrossRef] [PubMed]
- Flasche, S.; van Hoek, A.J.; Sheasby, E.; Waight, P.; Andrews, N.; Sheppard, C.; George, R.; Miller, E. Effect of pneumococcal conjugate vaccination on serotype-specific carriage and invasive disease in England: A cross-sectional study. PLoS Med. 2011, 8, 14. [Google Scholar] [CrossRef]
- van Hoek, A.J.; Sheppard, C.L.; Andrews, N.J.; Waight, P.A.; Slack, M.P.E.; Harrison, T.G.; Ladhani, S.N.; Miller, E. Pneumococcal carriage in children and adults two years after introduction of the thirteen valent pneumococcal conjugate vaccine in England. Vaccine 2014, 32, 4349–4355. [Google Scholar] [CrossRef] [PubMed]
- Southern, J.; Andrews, N.; Sandu, P.; Sheppard, C.L.; Waight, P.A.; Fry, N.K.; Van Hoek, A.J.; Miller, E. Pneumococcal carriage in children and their household contacts six years after introduction of the 13-valent pneumococcal conjugate vaccine in England. PLoS ONE 2018, 13, e0195799. [Google Scholar] [CrossRef]
- A Study to Assess the Carriage of Pneumococci in Children Aged ≤8 Years, and Their Household Contacts (PIN2). Available online: https://clinicaltrials.gov/ct2/show/NCT03407690?term=NCT03407690&rank=1 (accessed on 25 July 2019).
- Waight, P.A.; Andrews, N.J.; Ladhani, S.N.; Sheppard, C.L.; Slack, M.P.E.; Miller, E. Effect of the 13-valent pneumococcal conjugate vaccine on invasive pneumococcal disease in England and Wales 4 years after its introduction: An observational cohort study. Lancet Infect. Dis. 2015, 15, 535–543. [Google Scholar] [CrossRef]
- Ladhani, S.N.; Collins, S.; Djennad, A.; Sheppard, C.L.; Borrow, R.; Fry, N.K.; Andrews, N.J.; Miller, E.; Ramsay, M.E. Rapid increase in non-vaccine serotypes causing invasive pneumococcal disease in England and Wales, 2000–2017: A prospective national observational cohort study. Lancet Infect. Dis. 2018, 4, 441–451. [Google Scholar] [CrossRef]
- Chewapreecha, C.; Harris, S.R.; Croucher, N.J.; Turner, C.; Marttinen, P.; Cheng, L.; Pessia, A.; Aanensen, D.M.; Mather, A.E.; Page, A.J.; et al. Dense genomic sampling identifies highways of pneumococcal recombination. Nat. Genet. 2014, 46, 305–309. [Google Scholar] [CrossRef] [Green Version]
- Turner, P.; Turner, C.; Jankhot, A.; Helen, N.; Lee, S.J.; Day, N.P.; White, N.J.; Nosten, F.; Goldblatt, D. A Longitudinal Study of Streptococcus pneumoniae Carriage in a Cohort of Infants and Their Mothers on the Thailand-Myanmar Border. PLoS ONE 2012, 7, e38271. [Google Scholar] [CrossRef]
- Lees, J.A.; Croucher, N.J.; Goldblatt, D.; Nosten, F.; Parkhill, J.; Turner, C.; Turner, P.; Bentley, S.D. Genome-wide identification of lineage and locus specific variation associated with pneumococcal carriage duration. Elife 2017, 6, e26255. [Google Scholar] [CrossRef]
- Corander, J.; Fraser, C.; Gutmann, M.U.; Arnold, B.; Hanage, W.P.; Bentley, S.D.; Lipsitch, M.; Croucher, N.J. Frequency-dependent selection in vaccine-associated pneumococcal population dynamics. Nat. Ecol. Evol. 2017, 1, 1950. [Google Scholar] [CrossRef] [PubMed]
- Gladstone, R.A.; Jefferies, J.M.; Tocheva, A.S.; Beard, K.R.; Garley, D.; Chong, W.W.; Bentley, S.D.; Faust, S.N.; Clarke, S.C. Five winters of pneumococcal serotype replacement in UK carriage following PCV introduction. Vaccine 2015, 33, 17. [Google Scholar] [CrossRef] [PubMed]
- Tewolde, R.; Dallman, T.; Schaefer, U.; Sheppard, C.L.; Ashton, P.; Pichon, B.; Ellington, M.; Swift, C.; Green, J.; Underwood, A. MOST: A modified MLST typing tool based on short read sequencing. PeerJ 2016, 4, e2308. [Google Scholar] [CrossRef] [PubMed]
- Kapatai, G.; Sheppard, C.L.; Al-Shahib, A.; Litt, D.J.; Underwood, A.P.; Harrison, T.G.; Fry, N.K. Whole genome sequencing of Streptococcus pneumoniae: Development, evaluation and verification of targets for serogroup and serotype prediction using an automated pipeline. PeerJ 2016, 4, e2477. [Google Scholar] [CrossRef] [PubMed]
- Nascimento, M.; Sousa, A.; Ramirez, M.; Francisco, A.P.; Carriço, J.A.; Vaz, C. PHYLOViZ 2.0: Providing scalable data integration and visualization for multiple phylogenetic inference methods. Bioinformatics 2017, 33, 128–129. [Google Scholar] [CrossRef]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef] [PubMed]
- Lees, J.A.; Harris, S.R.; Tonkin-Hill, G.; Gladstone, R.A.; Lo, S.W.; Weiser, J.N.; Corander, J.; Bentley, S.D.; Croucher, N.J. Fast and flexible bacterial genomic epidemiology with PopPUNK. Genome Res. 2019, 29, 304–316. [Google Scholar] [CrossRef] [Green Version]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Abricate v0.8.13. Available online: https://github.com/ tseemann/abricate/ (accessed on 11 July 2019).
- Zankari, E.; Hasman, H.; Cosentino, S.; Vestergaard, M.; Rasmussen, S.; Lund, O.; Aarestrup, F.M.; Larsen, M.V. Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 2012, 67, 2640–2644. [Google Scholar] [CrossRef]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.G.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Smith, D.K.; Zhu, H.; Guan, Y.; Lam, T.T.-Y. ggtree: An r package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 2017, 8, 28–36. [Google Scholar] [CrossRef]
- Gladstone, R.A.; Lo, S.W.; Lees, J.A.; Croucher, N.J.; van Tonder, A.J.; Corander, J.; Page, A.J.; Marttinen, P.; Bentley, L.J.; Ochoa, T.J.; et al. International genomic definition of pneumococcal lineages, to contextualise disease, antibiotic resistance and vaccine impact. EBioMedicine 2019, 43, 338–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamada, T.; Kawai, S. An algorithm for drawing general undirected graphs. Inf. Process. Lett. 1989, 31, 7–15. [Google Scholar] [CrossRef]
- Shannon, P. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Makwana, A.; Ladhani, S.N.; Kapatai, G.; Campion, E.; Fry, N.K.; Sheppard, C. Rapid spread of pneumococcal nonvaccine serotype 7C previously associated with vaccine serotype 19F, England and Wales. Emerg. Infect. Dis. 2018, 24, 1919. [Google Scholar] [CrossRef]
- Lo, S.W.; Gladstone, R.A.; van Tonder, A.J.; Lees, J.A.; du Plessis, M.; Benisty, R.; Givon-Lavi, N.; Hawkins, P.A.; Cornick, J.E.; Kwambana-Adams, B.; et al. Pneumococcal lineages associated with serotype replacement and antibiotic resistance in childhood invasive pneumococcal disease in the post-PCV13 era: An international whole-genome sequencing study. Lancet Infect. Dis. 2019, 19, 759–769. [Google Scholar] [CrossRef]
- Andrews, N.; Kent, A.; Amin-Chowdhury, Z.; Sheppard, C.; Fry, N.; Ramsay, M.; Ladhani, S.N. Effectiveness of the seven-valent and thirteen-valent pneumococcal conjugate vaccines in England: The indirect cohort design, 2006–2018. Vaccine 2019, 37, 4491–4498. [Google Scholar] [CrossRef]
- Kapatai, G.; Sheppard, C.L.; Troxler, L.J.; Litt, D.J.; Furrer, J.; Hilty, M.; Fry, N.K. Pneumococcal 23B molecular subtype identified using whole genome sequencing. Genome Biol. Evol. 2017, 9, 2145–2158. [Google Scholar] [CrossRef]
- Azarian, T.; Mitchell, P.K.; Georgieva, M.; Thompson, C.M.; Ghouila, A.; Pollard, A.J.; von Gottberg, A.; du Plessis, M.; Antonio, M.; Kwambana-Adams, B.A.; et al. Global emergence and population dynamics of divergent serotype 3 CC180 pneumococci. PLoS Pathog. 2018, 14, e1007438. [Google Scholar] [CrossRef]
- Mitchell, P.K.; Azarian, T.; Croucher, N.J.; Callendrello, A.; Thompson, C.M.; Pelton, S.I.; Lipsitch, M.; Hanage, W.P. Population genomics of pneumococcal carriage in Massachusetts children following introduction of PCV-13. Microb. Genom. 2019, 5, e000252. [Google Scholar] [CrossRef] [PubMed]
- Jansen, K.U.; Anderson, A.S. The role of vaccines in fighting antimicrobial resistance (AMR). Hum. Vaccin. Immunother. 2018, 14, 2142–2149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obolski, U.; Lourenço, J.; Thompson, C.; Thompson, R.; Gori, A.; Gupta, S. Vaccination can drive an increase in frequencies of antibiotic resistance among nonvaccine serotypes of Streptococcus pneumoniae. Proc. Natl. Acad. Sci. USA 2018, 115, 3102–3107. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
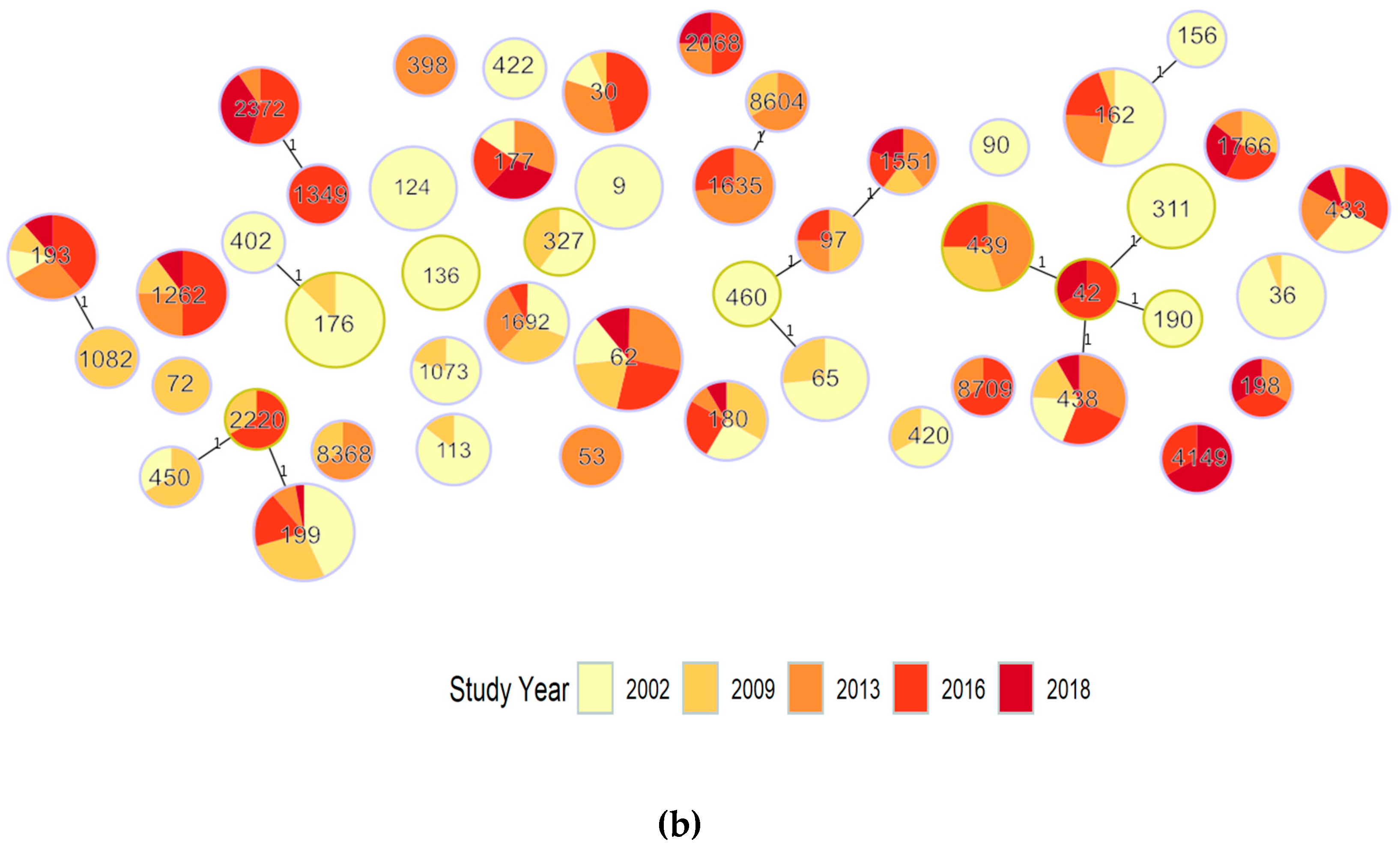{kind=link}
{kind=link}
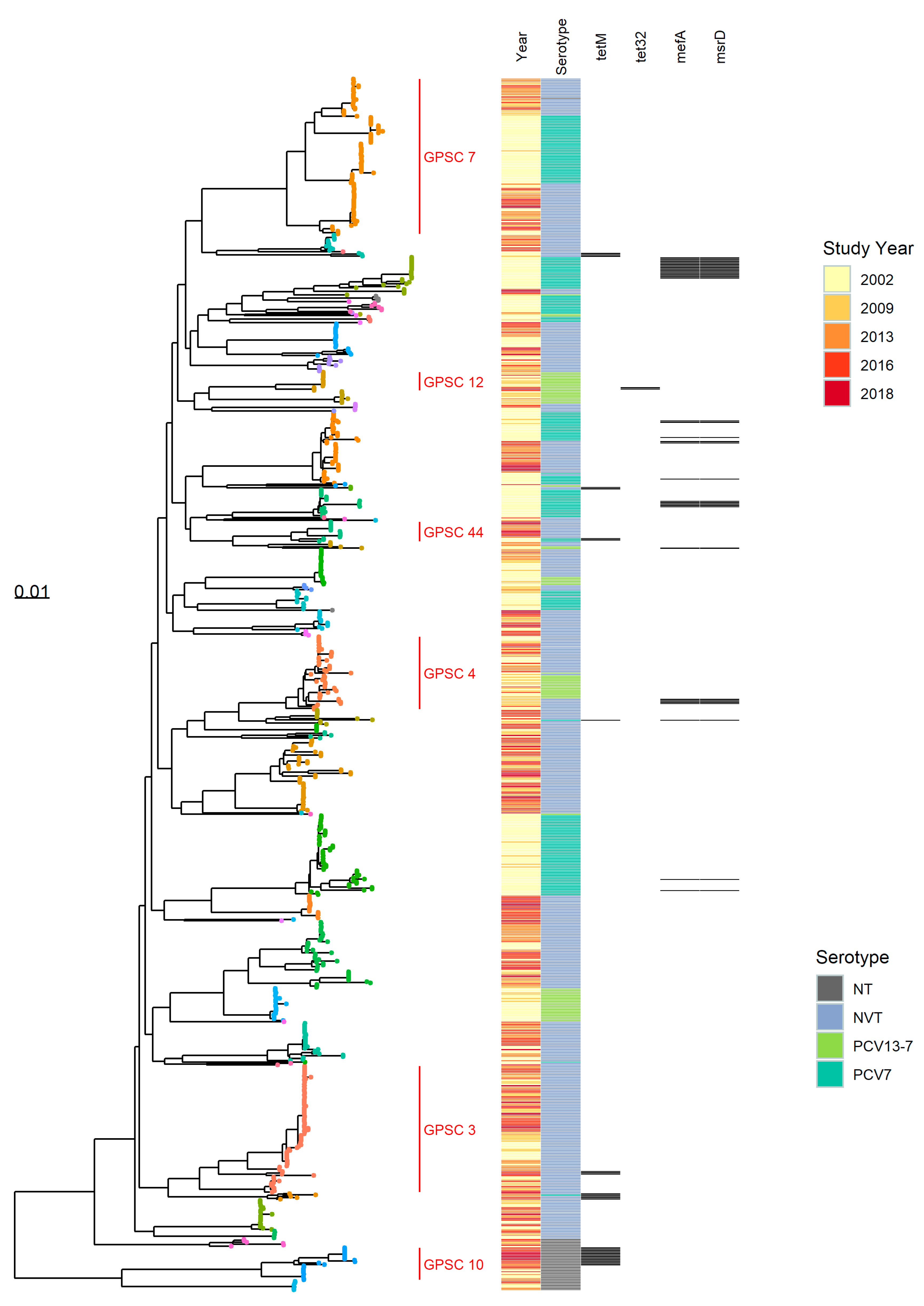{kind=link}
| Serotype Category | Number of Genomes per Year of Study | |||||
|---|---|---|---|---|---|---|
| 2002 | 2009 | 2013 | 2016 | 2018 | Total | |
| PCV7 | 162 | 10 | 2 | 0 | 0 | 174 |
| PCV13-7 | 36 | 23 | 2 | 5 | 1 | 67 |
| NVT | 80 | 66 | 119 | 143 | 48 | 456 |
| NT | 5 | 1 | 8 | 11 | 5 | 30 |
| Total | 283 | 100 | 131 | 160 | 54 | 727 |
| Time Period | No. STs | No. Genomes | No. of ST Represented by <3 Isolates | Genome to ST Ratio |
|---|---|---|---|---|
| Persisting Pre- and Post-PCV (2002 and any 2013-2018) | 25 | 343 | 1 | 13.7 |
| Pre-PCV7 (2002 only) | 50 | 117 | 43 | 2.34 |
| Pre-PCV13 (2002 and 2009 only) | 8 | 87 | 0 | 10.9 |
| Post-PCV7 (2009 and any 2013-2018 only) | 16 | 76 | 7 | 4.8 |
| Post-PCV13 (2013 and 2016 or 2018 only) | 51 | 104 | 38 | 2.0 |
| Total | 150 | 727 | 89 | - |
| Period No. ST, No. Genomes | ST | PopPUNK Cluster | GPSC Cluster | Serotype (Number of Genomes) | Total Genomes | ||||
|---|---|---|---|---|---|---|---|---|---|
| 2002 N = 283 | 2009 N = 100 | 2013 N = 131 | 2016 N = 160 | 2018 N = 54 | |||||
| Pre-PCV7 (2002 only) 50, 117 | 9 | 13 | 16 | 14 (16) | 16 | ||||
| 124 | 16 | 39 | 14 (15) | 15 | |||||
| 311 | 1 | 7 | 23F (15) | 15 | |||||
| 138 | 3 | 24 | 6B (8) | 8 | |||||
| 460 | 11 | 64 | 6A (4) | 4 | |||||
| 402 | 3 | 24 | 6B (3) | 3 | |||||
| 422 | 33 | 316 | 19F (3) | 3 | |||||
| 90 | 51 | 23 | 6B (2) | 2 | |||||
| 156 | 5 | 6 | 9V (2) | 2 | |||||
| 190 | 6 | 7 | 23A (2) | 2 | |||||
| Pre-PCV13 (2002 and 2009 only) 8, 87 | 176 | 3 | 24 | 6B (28) | 6B (4) | 32 | |||
| 36 | 1 | 7 | 23F (16) | 23F (1) | 17 | ||||
| 65 | 11 | 64 | 6A (11) | 6A (4) | 15 | ||||
| 113 | 21 | 50 | 18C (5) 18B (1) | 18C (1) | 7 | ||||
| 1073 | 21 | 50 | 18C (4) | 18C (1) | 5 | ||||
| 327 | 9 | 29 | 6A (3) | 6A (2) | 5 | ||||
| 420 | 44 | 1 | 19F (2) | 19F (1) | 3 | ||||
| 450 | 4 | 4 | 19A (1) | 19A (2) | 3 | ||||
| Post-PCV7 (2009 and any 2013–2018 only) 16, 76 | 439 | 1 | 7 | 23B (6) | 23B (8) NT (1) | 23B (5) | 20 | ||
| 1262 | 15 | 11 | 15B/C (3) | 15B/C (5) | 15B/C (10) | 15B/C (2) | 20 | ||
| 1766 | 22 | 57 | 31 (2) | 31 (1) | 31 (2) | 31 (2) | 7 | ||
| 1551 | 25 | 35 | 10A (1) | 10A (2) | 10A (1) | 10A (1) | 5 | ||
| 97 | 25 | 35 | 10A (2) | 10A (1) | 10A (1) | 4 | |||
| 1082 | 8 | 11 | 21 (3) | 3 | |||||
| 8368 | 26 | 99 | 21 (1) | 21 (2) | 3 | ||||
| 8604 | 12 | 36 | 35F (1) | 35F (2) | 3 | ||||
| 2220 | 4 | 4 | 15B/C (1) | 15B/C (2) | 3 | ||||
| 72 | 47 | 16 | 24 * (2) | 2 | |||||
| Post-PCV13 (2013 and 2016 or 2018 only) 51, 104 | 1635 | 12 | 36 | 35F (8) | 35F (3) | 11 | |||
| 2372 | 18 | 5 | 23B (1) | 23B (6) | 23B (4) | 11 | |||
| 4149 | 10 | 81 | NT (2) | NT (4) | 6 | ||||
| 2068 | 20 | 36 | 10A (1) | 10A (2) | 10A (1) | 4 | |||
| 53 | 48 | 3 | 8 (3) | 3 | |||||
| 398 | 49 | 13 | 6C (3) | 3 | |||||
| 8709 | 31 | 258 | NT (1) | NT (2) | 3 | ||||
| 198 | 34 | 72 | 35B (1) | 35B (1) | 35B (1) | 3 | |||
| 1349 | 18 | 5 | 23B (3) | 3 | |||||
| 42 | 6 | 7 | 23A (2) | 23A (1) | 3 | ||||
| Persisting pre-PCV7 to post-PCV13 (2002 and any 2013–2018) 25, 343 | 62 | 2 | 3 | 11A (9) | 11A (11) | 11A (16) | 11A (14) | 11A (6) | 56 |
| 162 | 5 | 6 | 19F (13) 9A (1) 9V (6) | 19F (2) | 24 * (7) 15B/C (1) | 24 * (4) 15B/C (3) | 37 | ||
| 199 | 4 | 4 | 19A (6) 15B/C (10) | 19A (5) 15B/C (5) | 15B/C (3) | 19A (2) 15B/C (5) | 15B/C (1) | 37 | |
| 438 | 6 | 7 | 23A (5) | 23A (4) | 23A (8) | 23A (6) | 23A (2) | 25 | |
| 433 | 14 | 19 | 22F (5) | 22F (1) | 22F (3) 42(1) | 22F (6) | 22F (2) | 18 | |
| 193 | 8 | 11 | 21 (2) | 21 (2) | 21 (5) | 21 (7) | 21 (2) | 18 | |
| 30 | 7 | 46 | 16F (2) | 16F (1) | 16F (5) | 16F (7) | 15 | ||
| 1692 | 9 | 29 | 6C (4) | 6C (4) | 6C (4) | 6C (1) | 13 | ||
| 177 | 19 | 44 | 19F (2) | 24 *(3) 7C (1) | 24 *(2) 7C (1) | 24 *(1) 7C (3) | 13 | ||
| 180 | 27, 46 | 12 | 3 (3) | 3 (4) | 3 (1) | 3 (3) | 3 (1) | 12 | |
| ST | % of Isolates per Study | Exact Test for Change between Years | Comment | ||||
|---|---|---|---|---|---|---|---|
| 2002 N = 283 | 2009 N = 100 | 2013 N = 131 | 2016 N = 160 | 2018 N = 54 | |||
| 9 | 5.7 | *** | Decrease post 2002 | ||||
| 124 | 5.3 | *** | Decrease post 2002 | ||||
| 311 | 5.3 | ** | Decrease post 2002 | ||||
| 176 | 9.9 | 4.0 | *** | Decrease post 2002 | |||
| 36 | 5.7 | 1.0 | *** | Decrease post 2002 | |||
| 65 | 3.9 | 4.0 | ** | Decrease post 2009 | |||
| 439 | 6.0 | 6.9 | 3.1 | *** | Increase post 2002 | ||
| 1262 | 3.0 | 3.8 | 6.2 | 3.7 | *** | Increase post 2002 | |
| 1635 | 7.4 | 1.9 | *** | Increase post 2009 | |||
| 2372 | 0.8 | 3.8 | 7.4 | *** | Increase post 2013 | ||
| 4149 | 1.3 | 7.4 | *** | Increase post 2013 | |||
| 3811 | 5.6 | *** | Increase post 2016 | ||||
| 62 | 3.2 | 11.0 | 12.2 | 8.8 | 11.1 | ** | Increase post 2009 |
| 177 | 0.7 | 3.1 | 1.9 | 7.4 | * | Increase post 2009 | |
| Resistance Marker | No. Genomes | Study Year (No. Genomes) | ||||
|---|---|---|---|---|---|---|
| 2002 N = 346 | 2009 N = 127 | 2013 N = 153 | 2016 N = 187 | 2018 N = 64 | ||
| msrD, mefA | 30 | 14(19), 19F(3), 6A(1), 6B(2), 9V(1) | 15B/C(3) | 15B/C (1) | ||
| tetM | 18 | 19F(1), 6B(2) | 6C(1) | 19F(1) NT(1) | 15A(3), 33F(2), NT(2) | 33F(1), NT(4) |
| tetM, msrD, mefA, cat(pC194) | 1 | 19F(1) | ||||
| tetM, cat(pC194) | 2 | 6B(2) | ||||
| tetM, aph(3′)-III | 2 | 15A(1) | NT(1) | |||
| tetM, ermB, aph(3′)-III | 2 | NT(2) | ||||
| tet32 | 2 | 3(1) | 3(1) | |||
| Total | 57 | 32 | 4 | 3 | 12 | 6 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sheppard, C.L.; Groves, N.; Andrews, N.; Litt, D.J.; Fry, N.K.; Southern, J.; Miller, E. The Genomics of Streptococcus pneumoniae Carriage Isolates from UK Children and Their Household Contacts, Pre-PCV7 to Post-PCV13. Genes 2019, 10, 687. https://doi.org/10.3390/genes10090687
Sheppard CL, Groves N, Andrews N, Litt DJ, Fry NK, Southern J, Miller E. The Genomics of Streptococcus pneumoniae Carriage Isolates from UK Children and Their Household Contacts, Pre-PCV7 to Post-PCV13. Genes. 2019; 10(9):687. https://doi.org/10.3390/genes10090687
Chicago/Turabian StyleSheppard, Carmen L., Natalie Groves, Nicholas Andrews, David J. Litt, Norman K. Fry, Jo Southern, and Elizabeth Miller. 2019. "The Genomics of Streptococcus pneumoniae Carriage Isolates from UK Children and Their Household Contacts, Pre-PCV7 to Post-PCV13" Genes 10, no. 9: 687. https://doi.org/10.3390/genes10090687
APA StyleSheppard, C. L., Groves, N., Andrews, N., Litt, D. J., Fry, N. K., Southern, J., & Miller, E. (2019). The Genomics of Streptococcus pneumoniae Carriage Isolates from UK Children and Their Household Contacts, Pre-PCV7 to Post-PCV13. Genes, 10(9), 687. https://doi.org/10.3390/genes10090687