NCBI’s Virus Discovery Hackathon: Engaging Research Communities to Identify Cloud Infrastructure Requirements

 , , , , , , , , ,
, , , , , , , , ,  , , , , and add
Show full author list
, , , , and add
Show full author list
Abstract
:1. Introduction
2. Materials and Methods
2.1. Participant Recruitment
2.2. Assembling Contigs from Metagenomic Data Sets
2.3. Megablast
2.4. Markov Clustering
2.5. Domain Mapping
2.6. VIGA
2.7. Machine Learning
3. Results
3.1. Hackathon Planning and Preparation
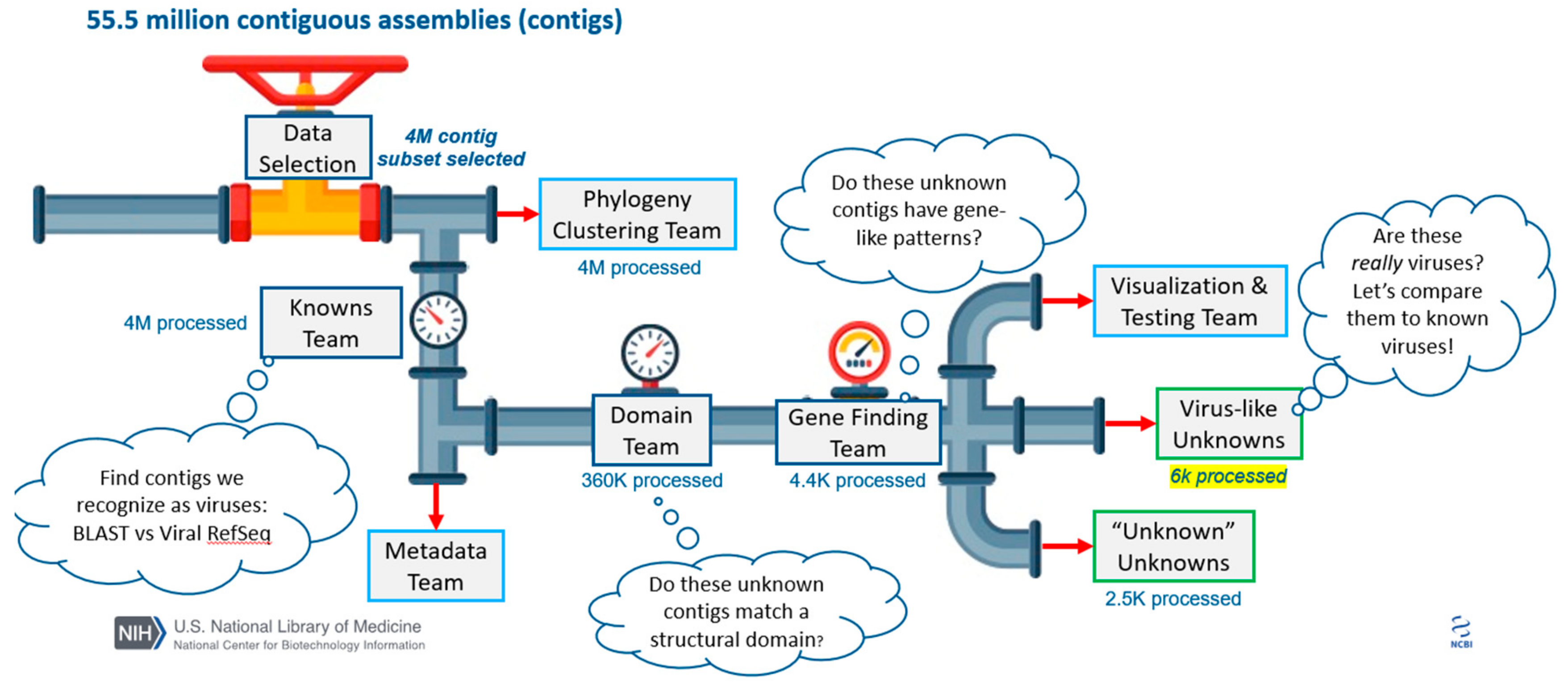
3.2. Data Selection
3.3. Data Segmentation
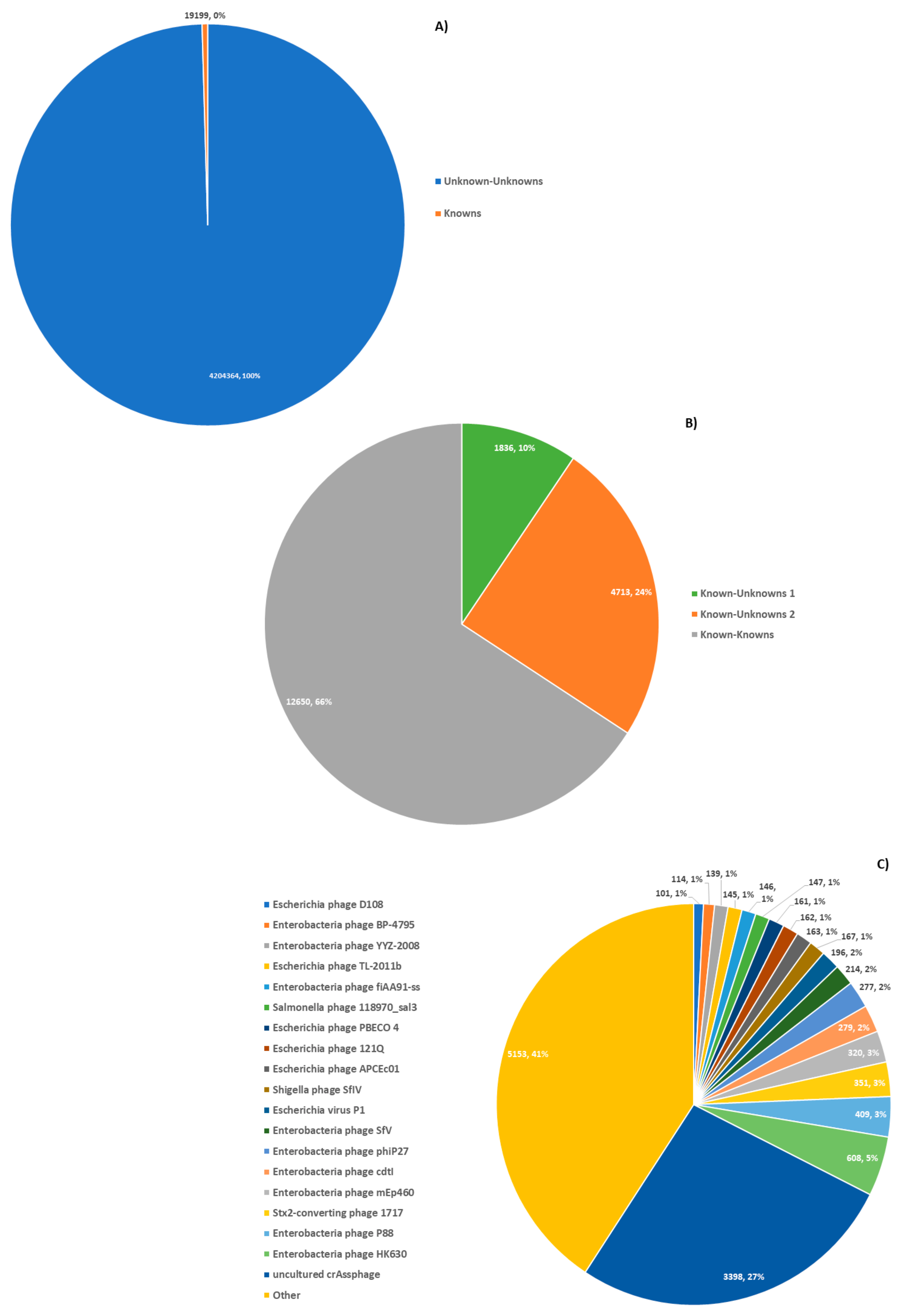
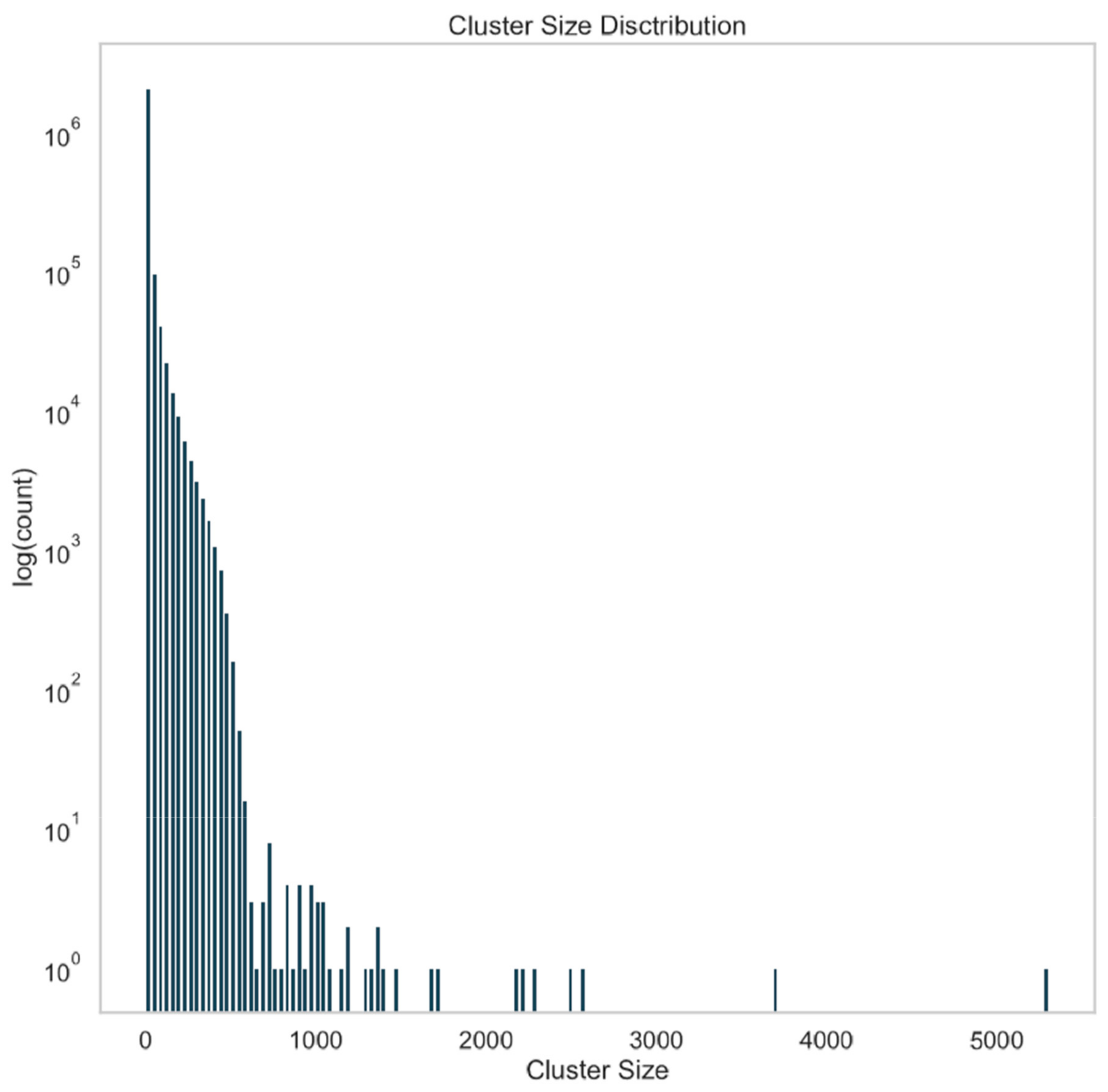
3.4. Data Clustering
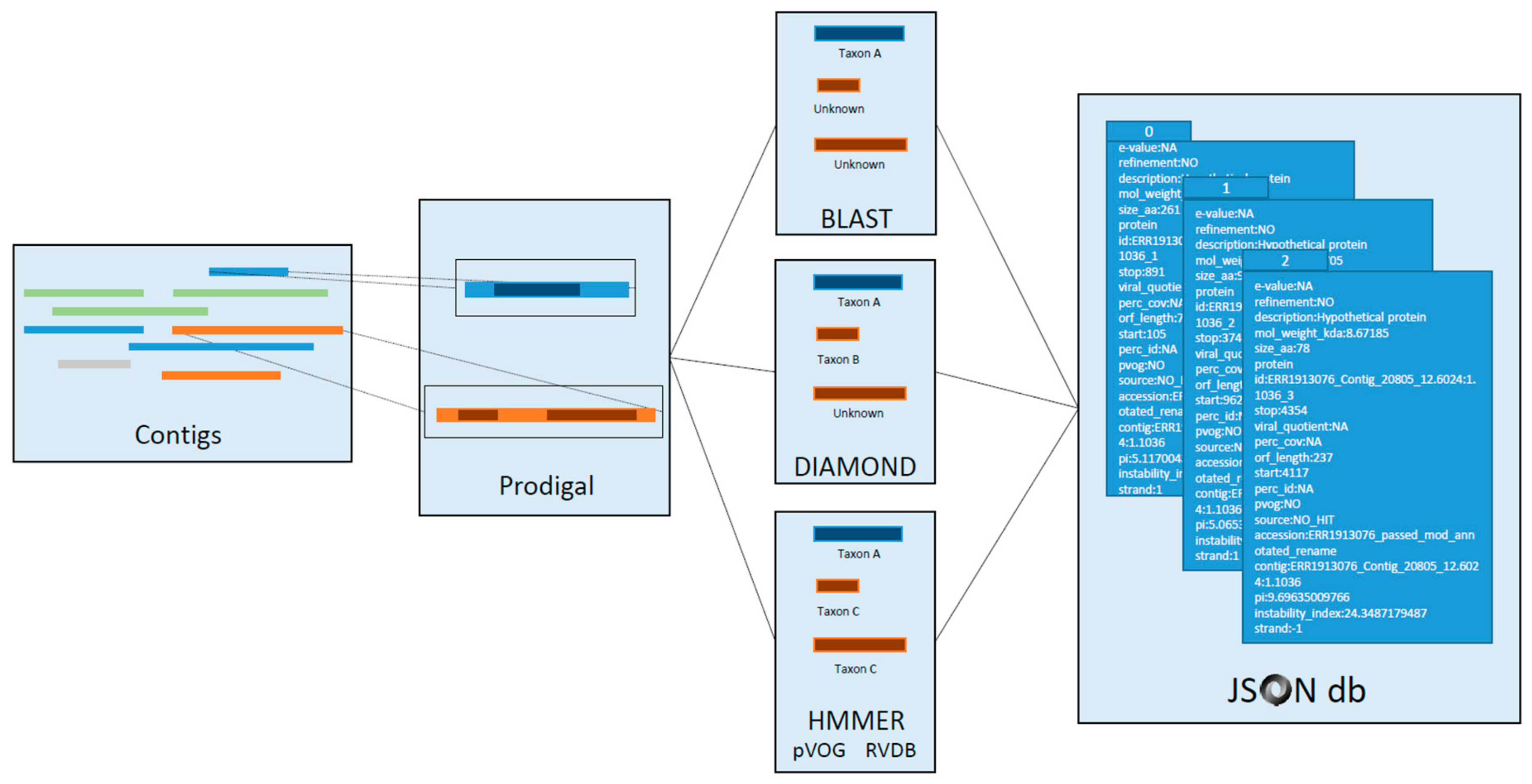
3.5. Domain Mapping
3.6. Gene Annotation
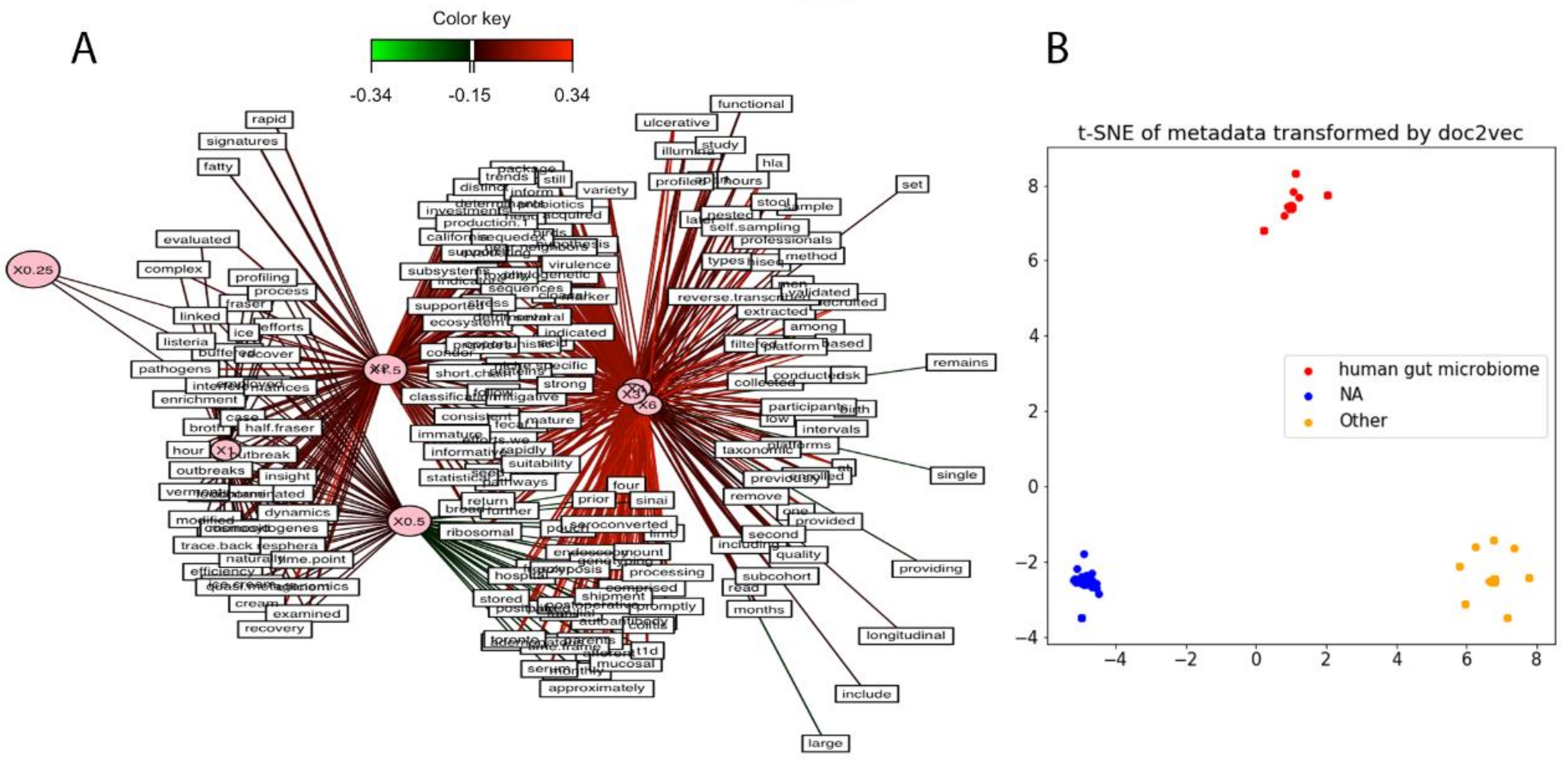
3.7. Metadata Analysis
4. Discussion
5. Conclusions
- Conservatively assembled contigs support initial exploration of SRA data.
- Redesigning algorithms to leverage cloud infrastructure would make cloud environments more accessible to a wider audience.
- Approaches to classifying—and reporting the classification of—contigs can be effectively developed via collaboration between a diverse group of researchers.
- Analysis will continue in a follow-up hackathon.
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mardis, E.R. A decade’s perspective on DNA sequencing technology. Nature 2011, 470, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Kodama, Y.; Shumway, M.; Leinonen, R. International Nucleotide Sequence Database Collaboration. The Sequence Read Archive: Explosive growth of sequencing data. Nucleic Acids Res. 2012, 40, D54–D56. [Google Scholar] [CrossRef] [PubMed]
- NIH Office of Data Science Strategy. STRIDES. 2019. Available online: https://datascience.nih.gov/strides (accessed on 15 July 2019).
- Sayers, E.W.; Agarwala, R.; Bolton, E.E.; Brister, J.R.; Canese, K.; Clark, K.; Connor, R.; Fiorini, N.; Funk, K.; Hefferon, T.; et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2019, 47, D23–D28. [Google Scholar] [CrossRef] [PubMed]
- Harkut, D.G.; Kasat, K.; Shah, S. Cloud Computing: Technology and Practices; BoD–Books on Demand: Norderstedt, Germany, 2019. [Google Scholar]
- Leinonen, R.; Sugawara, H.; Shumway, M. International Nucleotide Sequence Database Collaboration. The Sequence Read Archive. Nucleic Acids Res. 2010, 39, D19–D21. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Hallam, S.J.; Woyke, T.; Sullivan, M.B. Viral dark matter and virus-host interactions resolved from publicly available microbial genomes. eLife 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Carroll, D.; Daszak, P.; Wolfe, N.D.; Gao, G.F.; Morel, C.M.; Morzaria, S.; Pablos-Méndez, A.; Tomori, O.; Mazet, J.A.K. The Global Virome Project. Science 2018, 359, 872–874. [Google Scholar] [CrossRef]
- Torres, P.J.; Edwards, R.A.; McNair, K.A. PARTIE: A partition engine to separate metagenomic andamplicon projects in the Sequence Read Archive. Bioinformatics 2017, 33, 2389–2391. [Google Scholar] [CrossRef]
- NCBI-Hackathons/VirusDiscoveryProject. Available online: https://github.com/NCBI-Hackathons/VirusDiscoveryProject/blob/master/DataSelection/hackathon.sets (accessed on 12 September 2019).
- Souvorov, A.; Agarwala, R.; Lipman, D.J. SKESA: Strategic k-mer extension for scrupulous assemblies. Genome Biol. 2018, 19, 153. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- NCBI-Hackathons/VirusDiscoveryProject. Available online: https://github.com/NCBI-Hackathons/VirusDiscoveryProject (accessed on 12 September 2019).
- Ondov, B.D.; Starrett, G.J.; Sappington, A.; Kostic, A.; Koren, S.; Buck, C.B.; Phillippy, A.M. Mash Screen: High-throughput sequence containment estimation for genome discovery. BioRxiv 2019, 557314. [Google Scholar] [CrossRef]
- Hulo, C.; De Castro, E.; Masson, P.; Bougueleret, L.; Bairoch, A.; Xenarios, I.; Le Mercier, P. ViralZone: A knowledge resource to understand virus diversity. Nucleic Acids Res. 2011, 39, D576–D582. [Google Scholar] [CrossRef]
- NCBI Entrez Nucleotide database. Available online: https://www.ncbi.nlm.nih.gov/labs/virus/vssi/#/virus?VirusLineage_ss=Viruses,%20taxid:10239&SeqType_s=Nucleotide (accessed on 12 September 2019).
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Enright, A.J.; Van Dongen, S.; Ouzounis, C.A. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002, 30, 1575–1584. [Google Scholar] [CrossRef]
- Marchler-Bauer, A.; Bo, Y.; Han, L.; He, J.; Lanczycki, C.J.; Lu, S.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; et al. CDD/SPARCLE: Functional classification of proteins via subfamily domain architectures. Nucleic Acids Res. 2017, 45, D200–D203. [Google Scholar] [CrossRef]
- VirusDiscoveryProject/DomainLabeling/example. Available online: https://github.com/NCBI-Hackathons/VirusDiscoveryProject/tree/master/DomainLabeling/example (accessed on 12 September 2019).
- González-Tortuero, E.; Sutton, T.D.; Velayudhan, V.; Shkoporov, A.N.; Draper, L.A.; Stockdale, S.R.; Ross, R.P.; Hill, C. VIGA: A sensitive, precise and automatic de novo VIral Genome Annotator. BioRxiv 2018, 277509. [Google Scholar] [CrossRef]
- Nawrocki, E.P.; Eddy, S.R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 2013, 29, 2933–2935. [Google Scholar] [CrossRef] [Green Version]
- Eddy, S.R. HMMER: Biosequence Analysis Using Profile Hidden Markov Models. Available online: http://hmmer.org (accessed on 15 July 2019).
- Grazziotin, A.L.; Koonin, E.V.; Kristensen, D.M. Prokaryotic Virus Orthologous Groups (pVOGs): A resource for comparative genomics and protein family annotation. Nucleic Acids Res. 2017, 45, D491–D498. [Google Scholar] [CrossRef]
- Goodacre, N.; Aljanahi, A.; Nandakumar, S.; Mikailov, M.; Khan, A.S. A Reference Viral Database (RVDB) lsTo Enhance Bioinformatics Analysis of High-Throughput Sequencing for Novel Virus Detection. mSphere 2018, 3. [Google Scholar] [CrossRef]
- VirusDiscoveryProject/VirusGenes. Available online: https://github.com/NCBI-Hackathons/VirusDiscoveryProject/tree/master/VirusGenes (accessed on 12 September 2019).
- Choi, I.; Ponsero, A.J.; Bomhoff, M.; Youens-Clark, K.; Hartman, J.H.; Hurwitz, B.L. Libra: Scalable k-mer-based tool for massive all-vs-all metagenome Comparisons. GigaScience 2018, 8. [Google Scholar] [CrossRef]
- Python package scikit-learn. Available online: https://scikit-learn.org/stable/modules/generated/sklearn.manifold.TSNE.html (accessed on 12 September 2019).
- The number of metagenomic data sets in the SRA database. Available online: https://www.ncbi.nlm.nih.gov/sra/docs/sragrowth (accessed on 12 September 2019).
- Shi, M.; Lin, X.D.; Tian, J.H.; Chen, L.J.; Chen, X.; Li, C.X.; Qin, X.C.; Li, J.; Cao, J.P.; Eden, J.S.; et al. Redefining the invertebrate RNA virosphere. Nature 2016, 540, 539–543. [Google Scholar] [CrossRef]
- Jupyter Steering Council. The Jupyter/IPython Project. Available online: https://jupyter.org (accessed on 15 July 2019).
- Brister, J.R.; Ako-Adjei, D.; Bao, Y.; Blinkova, O. NCBI viral genomes resource. Nucleic Acids Res. 2015, 43, D571–D577. [Google Scholar] [CrossRef]
- Dutilh, B.E.; Cassman, N.; McNair, K.; Sanchez, S.E.; Silva, G.G.Z.; Boling, L.; Barr, J.J.; Speth, D.R.; Seguritan, V.; Aziz, R.K.; et al. A highly abundant bacteriophage discovered in the unknown sequences of human faecal metagenomes. Nat. Commun. 2014, 5, 4498. [Google Scholar] [CrossRef]
- Roux, S.; Enault, F.; Hurwitz, B.L.; Sullivan, M.B. VirSorter: Mining viral signal from microbial genomic data. PeerJ 2015, 3, e985. [Google Scholar] [CrossRef]
- Bray, T. The JavaScript Object Notation (JSON) Data Interchange Format. RFC 7159. RFC Editor, Ed.; Available online: https://www.rfc-editor.org (accessed on 12 August 2019).
- Hyatt, D.; Chen, G.L.; Locascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Zhu, Y.; Stephens, R.M.; Meltzer, P.S.; Davis, S.R. SRAdb: Query and use public next-generation sequencing data from within R. BMC Bioinform. 2013, 14, 19. [Google Scholar] [CrossRef]
- Le, Q.V.; Mikolov, T. Distributed Representations of Sentences and Documents. arXiv 2014, arXiv:1405.4053v2. [Google Scholar]
- Van der Maaten, L.; Hinton, G. Visualizing Data using t-SNE. J. Mach. Learn. Res. 2008, 9, 2579–2605. [Google Scholar]
- Wilkinson, M.D.; Dumontier, M.; Aalbersberg, I.J.J.; Appleton, G.; Axton, M.; Baak, A.; Blomberg, N.; Boiten, J.W.; Da Silva Santos, L.B.; Bourne, P.E.; et al. The FAIR Guiding Principles for scientific data management and stewardship. Sci. Data 2016, 3, 160018. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Raw | % Participants | % Responses | |
|---|---|---|---|
| Participants | 37 | NA | NA |
| Survey Responses | 36 | 97.3 | NA |
| Institutional Affiliation | |||
| Academic | 27 | 72.97 | 75.00 |
| Government | 6 | 16.22 | 16.67 |
| Other | 2 | 5.41 | 5.56 |
| Unknown | 1 | 2.70 | 2.80 |
| Educational Attainment | |||
| Ph.D. | 14 | 37.84 | 38.89 |
| M.S. | 12 | 32.43 | 33.33 |
| B.S. | 2 | 5.41 | 5.56 |
| Unknown | 8 | 21.62 | 22.22 |
| Career Stage | |||
| In Training | 11 | 29.73 | 30.56 |
| Junior | 14 | 37.84 | 38.89 |
| Senior | 6 | 16.22 | 16.67 |
| Unknown | 5 | 13.51 | 13.89 |
| Programming Language | |||
| Shell | 33 | 89.19 | 91.67 |
| Python | 31 | 83.78 | 86.11 |
| R | 26 | 70.27 | 72.22 |
| Perl | 13 | 35.14 | 36.11 |
| Java | 10 | 27.03 | 27.78 |
| C/C++ | 9 | 24.32 | 25.00 |
| JavaScript | 4 | 10.81 | 11.11 |
| SQL | 3 | 8.11 | 8.33 |
| Matlab | 2 | 5.41 | 5.56 |
| Other | 4 | 10.81 | 11.11 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Connor, R.; Brister, R.; Buchmann, J.P.; Deboutte, W.; Edwards, R.; Martí-Carreras, J.; Tisza, M.; Zalunin, V.; Andrade-Martínez, J.; Cantu, A.; et al. NCBI’s Virus Discovery Hackathon: Engaging Research Communities to Identify Cloud Infrastructure Requirements. Genes 2019, 10, 714. https://doi.org/10.3390/genes10090714
Connor R, Brister R, Buchmann JP, Deboutte W, Edwards R, Martí-Carreras J, Tisza M, Zalunin V, Andrade-Martínez J, Cantu A, et al. NCBI’s Virus Discovery Hackathon: Engaging Research Communities to Identify Cloud Infrastructure Requirements. Genes. 2019; 10(9):714. https://doi.org/10.3390/genes10090714
Chicago/Turabian StyleConnor, Ryan, Rodney Brister, Jan P. Buchmann, Ward Deboutte, Rob Edwards, Joan Martí-Carreras, Mike Tisza, Vadim Zalunin, Juan Andrade-Martínez, Adrian Cantu, and et al. 2019. "NCBI’s Virus Discovery Hackathon: Engaging Research Communities to Identify Cloud Infrastructure Requirements" Genes 10, no. 9: 714. https://doi.org/10.3390/genes10090714
APA StyleConnor, R., Brister, R., Buchmann, J. P., Deboutte, W., Edwards, R., Martí-Carreras, J., Tisza, M., Zalunin, V., Andrade-Martínez, J., Cantu, A., D’Amour, M., Efremov, A., Fleischmann, L., Forero-Junco, L., Garmaeva, S., Giluso, M., Glickman, C., Henderson, M., Kellman, B., ... Busby, B. (2019). NCBI’s Virus Discovery Hackathon: Engaging Research Communities to Identify Cloud Infrastructure Requirements. Genes, 10(9), 714. https://doi.org/10.3390/genes10090714