Genetic Analysis of QTL for Resistance to Maize Lethal Necrosis in Multiple Mapping Populations

 ,
,  ,
, 

Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Phenotypic Evaluation
2.3. Phenotypic Data Analysis
2.4. DNA Extraction, Genotyping, Linkage Map Construction and QTL Analysis
2.5. Joint Linkage Association Mapping (JLAM)
2.6. Genomic Prediction
3. Results
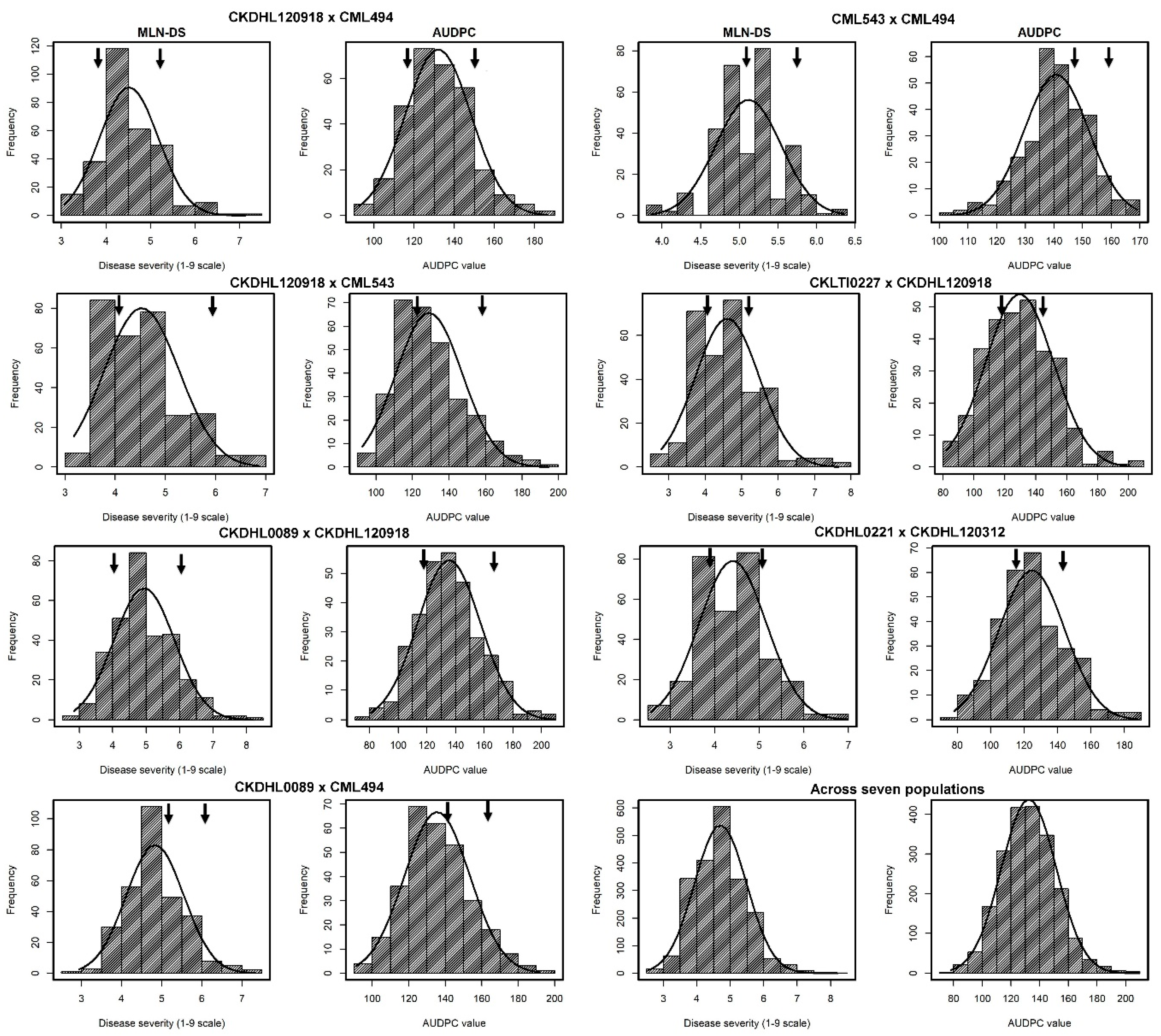
3.1. Response of Parents and F3 Populations to MLN Infections
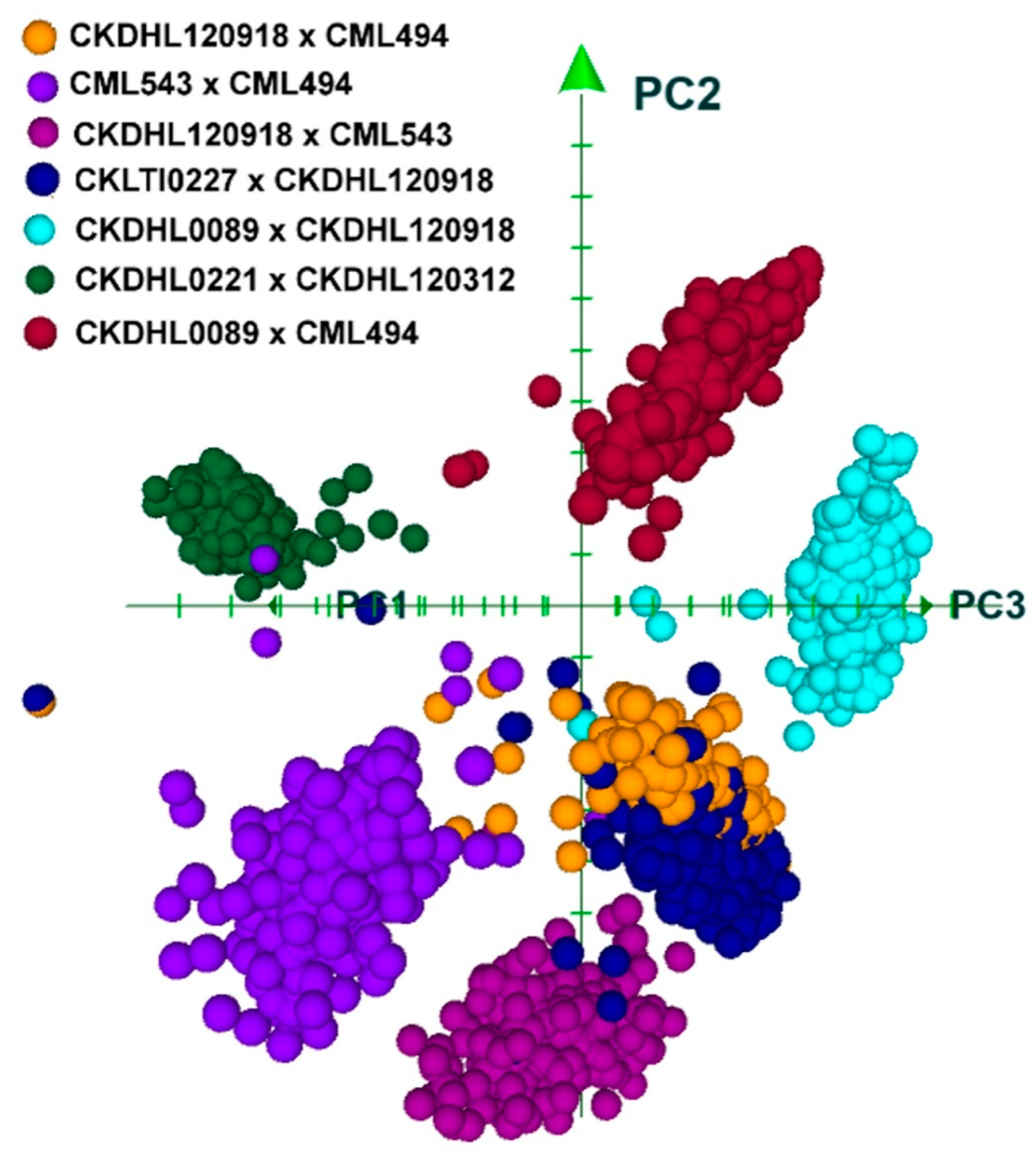
3.2. Molecular Analyses
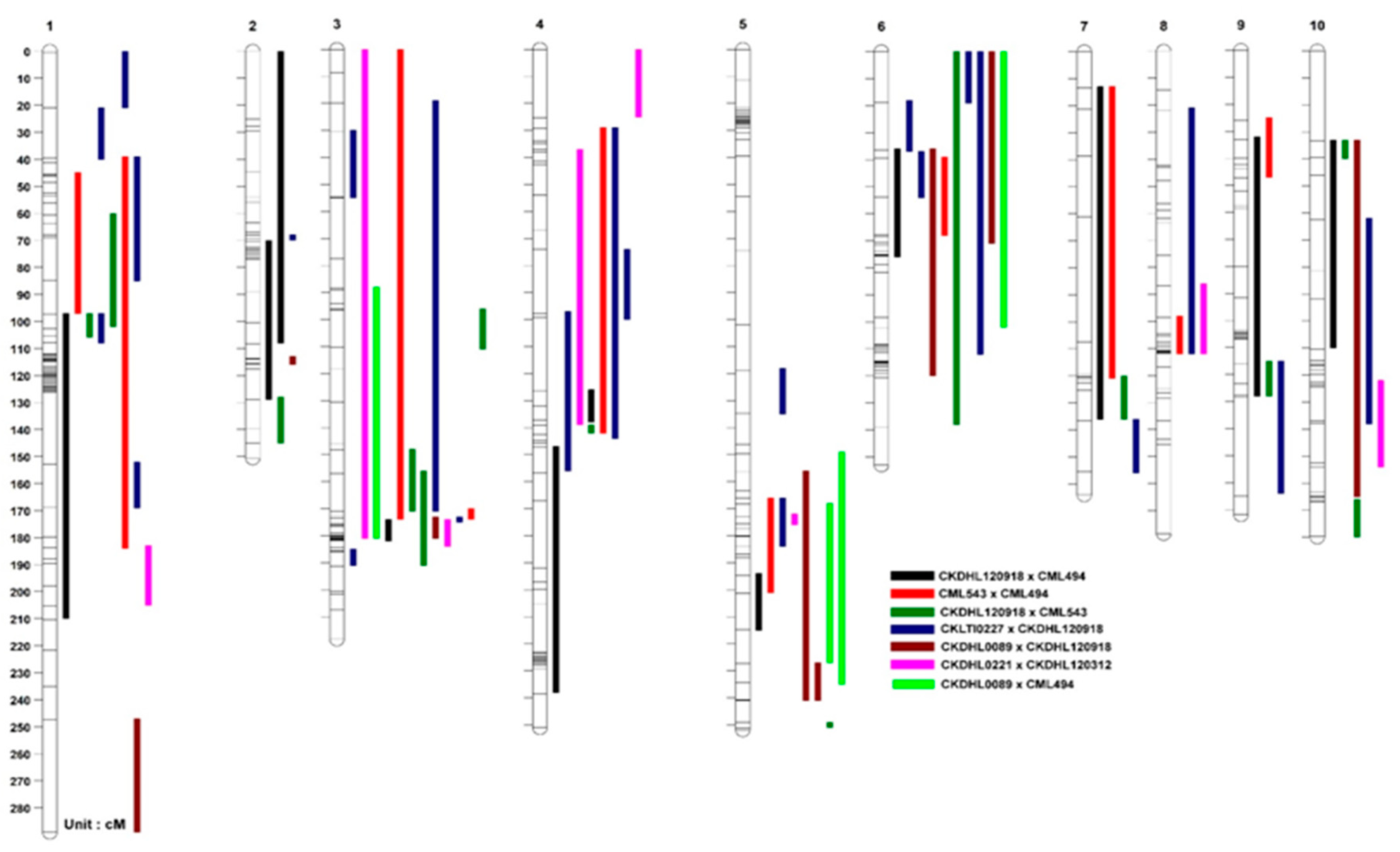
3.2.1. Linkage Group
3.2.2. QTL Mapping
3.2.3. Consensus Map Construction
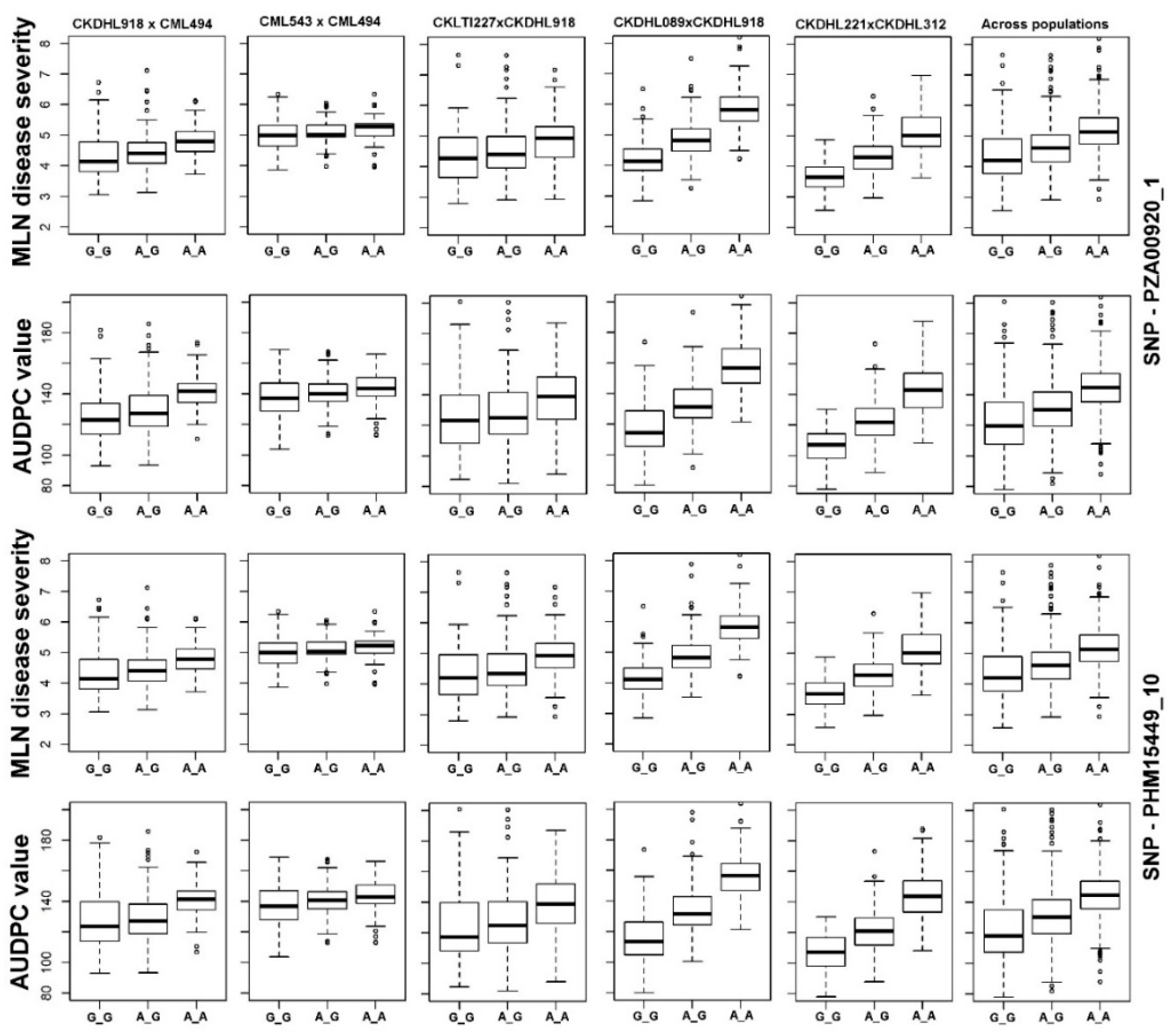
3.2.4. Joint Linkage Association Mapping (JLAM)
4. Discussion
4.1. Response of Parents and F3 Populations to MLN Infections
4.2. QTL Analyses
4.3. Joint Linkage Association Mapping (JLAM)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mahuku, G.; Lockhart, B.E.; Wanjala, B.; Jones, M.W.; Kimunye, J.N.; Stewart, L.R.; Cassone, B.J.; Subramanian, S.; Nyasani, J.; Kusia, E.; et al. Maize lethal necrosis (MLN), an emerging threat to maize-based food security in sub-Saharan Africa. Phytopathol. 2015, 105, 956–965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wamaitha, M.J.; Nigam, D.; Maina, S.; Stomeo, F.; Wangai, A.; Njuguna, J.N.; Holton, T.A.; Wanjala, B.W.; Wamalwa, M.; Lucas, T.; et al. Metagenomic analysis of viruses associated with maize lethal necrosis in Kenya. Virol. J. 2018, 15, 90. [Google Scholar] [CrossRef] [PubMed]
- Wangai, A.W.; Redinbaugh, M.G.; Kinyua, Z.M.; Miano, D.W.; Leley, P.K.; Kasina, M.; Mahuku, G.; Scheets, K.; Jeffers, D. First Report of Maize chlorotic mottle virus and Maize Lethal Necrosis in Kenya. Plant Dis. 2012, 96, 1582. [Google Scholar] [CrossRef] [PubMed]
- Redinbaugh, M.; Pratt, R. Virus resistance. In Handbook of Maize: It’s Biology; Bennetzen, J.L., Hake, S.C., Eds.; Springer: New York, NY, USA, 2009; pp. 251–268. [Google Scholar]
- Bonamico, N.C.; Di Renzzo, M.; Ibanez, M.; Borghi, M.; Diaz, D.; Salerno, J.; Balzarini, M. QTL analysis of resistance to Mal de Río Cuarto disease in maize using recombinant inbred lines QTL analysis of resistance to Mal de Río Cuarto disease in maize using recombinant inbred lines. J. Agric. Sci. 2012, 150, 5. [Google Scholar] [CrossRef]
- Zambrano, J.L.; Jones, M.W.; Brenner, E.; Francis, D.M.; Tomas, A.; Redinbaugh, M.G. Genetic analysis of resistance to six virus diseases in a multiple virus-resistant maize inbred line. Theor. Appl. Genet. 2014, 127, 867–880. [Google Scholar] [CrossRef]
- Gowda, M.; Beyene, Y.; Makumbi, D.; Semagn, K.; Olsen, M.S.; Bright, J.M.; Das, B.; Mugo, S.; Suresh, L.M.; Prasanna, B.M. Discovery and validation of genomic regions associated with resistance to maize lethal necrosis in four biparental populations. Mol. Breed. 2018, 38, 66. [Google Scholar] [CrossRef] [Green Version]
- Sitonik, C.; Suresh, L.M.; Beyene, Y.; Olsen, M.S.; Makumbi, D.; Oliver, K.; Das, B.; Bright, J.M.; Mugo, S.; Crossa, J.; et al. Genetic architecture of maize chlorotic mottle virus and maize lethal necrosis through GWAS, linkage analysis and genomic prediction in tropical maize germplasm. Theor. Appl. Genet. 2019, 132, 2381–2399. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Holland, J.B.; McMullen, M.D.; Buckler, E.S. Genetic Design and Statistical Power of Nested Association Mapping in Maize. Genet 2008, 178, 539–551. [Google Scholar] [CrossRef] [Green Version]
- Wurschum, T.; Liu, W.; Gowda, M.; Maurer, H.P.; Fischer, S.; Schechert, A.; Reif, J.C. Comparison of biometrical models for joint linkage association mapping. Heredity 2012, 108, 332–340. [Google Scholar] [CrossRef] [Green Version]
- Hawkins, L.K.; Warburton, M.L.; Tang, J.D.; Tomashek, J.; Oliveira, D.A.; Ogunola, O.F.; Smith, J.S.; Williams, W.P. Survey of Candidate Genes for Maize Resistance to Infection by Aspergillus flavus and/or Aflatoxin Contamination. Toxins 2018, 10, 61. [Google Scholar] [CrossRef] [Green Version]
- Gelli, M.; Konda, A.R.; Liu, K.; Zhang, C.; Clemente, T.E.; Holding, D.R.; Dweikat, I.M. Validation of QTL mapping and transcriptome profiling for identification of candidate genes associated with nitrogen stress tolerance in sorghum. BMC Plant Boil. 2017, 17, 123. [Google Scholar] [CrossRef] [PubMed]
- Sudheesh, S.; Rodda, M.S.; Davidson, J.; Javid, M.; Stephens, A.; Slater, A.T.; Cogan, N.O.I.; Forster, J.W.; Kaur, S. SNP-Based Linkage Mapping for Validation of QTLs for Resistance to Ascochyta Blight in Lentil. Front. Plant Sci. 2016, 7, 311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gajjar, K.N.; Mishra, G.P.; Radhakrishnan, T.; Dodia, S.M.; Rathnakumar, A.L.; Kumar, N.; Kumar, K.; Dobaria, J.R.; Kumar, A. Validation of SSR markers linked to the rust and late leaf spot diseases resistance in diverse peanut genotypes. Aust. J. Crop. Sci. 2014, 8, 927–936. [Google Scholar]
- Landi, P.; Sanguineti, M.C.; Salvi, S.; Giuliani, S.; Bellotti, M.; Maccaferri, M.; Conti, S.; Tuberosa, R. Validation and characterization of a major QTL affecting leaf ABA concentration in maize. Mol. Breed. 2005, 15, 291–303. [Google Scholar] [CrossRef]
- Sukruth, M.; Paratwagh, S.A.; Sujay, V.; Kumari, V.; Gowda, M.V.C.; Nadaf, H.L.; Motagi, B.N.; Lingaraju, S.; Pandey, M.K.; Varshney, R.K.; et al. Validation of markers linked to late leaf spot and rust resistance, and selection of superior genotypes among diverse recombinant inbred lines and backcross lines in peanut (Arachis hypogaea L.). Euphytica 2015, 204, 343–351. [Google Scholar] [CrossRef]
- Zhou, H.; Liu, S.; Liu, Y.; Liu, Y.; You, J.; Deng, M.; Ma, J.; Chen, G.; Wei, Y.; Liu, C.; et al. Mapping and validation of major quantitative trait loci for kernel length in wild barley (Hordeum vulgare ssp. spontaneum). BMC Genet. 2016, 17, 130. [Google Scholar] [CrossRef] [Green Version]
- De Leon, T.B.; Linscombe, S.; Subudhi, P.K. Identification and validation of QTLs for seedling salinity tolerance in introgression lines of a salt tolerant rice landrace ‘Pokkali’. PLoS ONE 2017, 12, e0175361. [Google Scholar] [CrossRef]
- Ishikawa, G.; Nakamura, K.; Ito, H.; Saito, M.; Sato, M.; Jinno, H.; Yoshimura, Y.; Nishimura, T.; Maejima, H.; Uehara, Y.; et al. Association Mapping and Validation of QTLs for Flour Yield in the Soft Winter Wheat Variety Kitahonami. PLoS ONE 2014, 9, e111337. [Google Scholar] [CrossRef]
- Bernardo, R. Molecular Markers and Selection for Complex Traits in Plants: Learning from the Last 20 Years. Crop. Sci. 2008, 48, 1649. [Google Scholar] [CrossRef] [Green Version]
- Tuberosa, R.; Salvi, S.; Sanguineti, M.C.; Landi, P.; Maccaferri, M.; Conti, S. Mapping QTLs Regulating Morpho-physiological Traits and Yield: Case Studies, Shortcomings and Perspectives in Drought-stressed Maize. Ann. Bot. 2002, 89, 941–963. [Google Scholar] [CrossRef] [Green Version]
- Gowda, M.; Das, B.; Makumbi, D.; Babu, R.; Semagn, K.; Mahuku, G.; Olsen, M.S.; Bright, J.M.; Beyene, Y.; Prasanna, B.M. Genome-wide association and genomic prediction of resistance to maize lethal necrosis disease in tropical maize germplasm. Theor. Appl. Genet. 2015, 128, 1957–1968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tivoli, B.; Baranger, A.; Avila, C.M.; Banniza, S.; Barbetti, M.; Chen, W.; Davidson, J.; Lindeck, K.; Kharrat, M.; Rubiales, D.; et al. Screening techniques and sources of resistance to foliar diseases caused by major necrotrophic fungi in grain legumes. Euphytica 2006, 147, 223–253. [Google Scholar] [CrossRef]
- Wang, J.; Li, H.; Zhang, L.; Meng, L. Users’ Manual of QTL IciMapping, 4.1; International Maize and Wheat Improvement Center (CIMMYT): Texcoco, Mexico, 2016. [Google Scholar]
- Alvarado, G.; López, M.; Vargas, M.; Pacheco, A.; Rodriguez, F.; Burgueno, J.; Crossa, J. META-R (Multi Environment Trail Analysis with R for Windows) Version 5.0—CIMMYT Research Software Dataverse—CIMMYT Dataverse Network. 2015. Available online: http://hdl.handle.net/11529/10201 (accessed on 5 September 2019).
- Stram, D.O.; Lee, J.W. Variance Components Testing in the Longitudinal Mixed Effects Model. Biometrics 1994, 50, 1171–1177. [Google Scholar] [CrossRef] [PubMed]
- CIMMYT. Laboratoty Protocols: CIMMYT Applied and Molecular Genetics Laboratoty, 3rd ed.; CIMMYT: Batan, Mexico, 2005. [Google Scholar]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef]
- Xia, X.; Melchinger, A.E.; Kuntze, L.; Lübberstedt, T. Quantitative Trait Loci Mapping of Resistance to Sugarcane Mosaic Virus in Maize. Phytopathology 1999, 89, 660–667. [Google Scholar] [CrossRef] [Green Version]
- Horn, F.; Habekuss, A.; Stich, B.; Habekuß, A. Linkage mapping of Barley yellow dwarf virus resistance in connected populations of maize. BMC Plant Boil. 2015, 15, 29. [Google Scholar] [CrossRef] [Green Version]
- Meng, L.; Li, H.; Zhang, L.; Wang, J. QTL IciMapping: Integrated software for genetic linkage map construction and quantitative trait locus mapping in biparental populations. Crop. J. 2015, 3, 269–283. [Google Scholar] [CrossRef] [Green Version]
- Kosambi, D. The estimation of map distances from recombination values. Ann. Eugen. 1944, 12, 172–175. [Google Scholar] [CrossRef]
- Voorrips, R.E. MapChart: Software for the graphical presentation of linkage maps and QTLs. J. Hered. 2002, 93, 77–78. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Ye, G.; Wang, J. A modified algorithm for the improvement of composite interval mapping. Genetics 2007, 175, 361–374. [Google Scholar] [CrossRef] [Green Version]
- Lübberstedt, T.; Melchinger, A.E.; Schön, C.C.; Utz, H.F.; Klein, D. QTL Mapping in Testcrosses of European Flint Lines of Maize: I. Comparison of Different Testers for Forage Yield Traits. Crop. Sci. 1997, 37, 921. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, L.; Fu, N.; Lou, Q.; Mei, H.; Xiong, L.; Li, M.; Xu, X.; Mei, X.; Luo, L. Dissection of additive, epistatic effect and QTL × environment interaction of quantitative trait loci for sheath blight resistance in rice. Hereditas 2014, 151, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Stuber, C.W.; Edwards, M.D.; Wendel, J.F. Molecular Marker-Facilitated Investigations of Quantitative Trait Loci in Maize. II. Factors Influencing Yield and its Component Traits1. Crop. Sci. 1987, 27, 639. [Google Scholar] [CrossRef] [Green Version]
- Carlson, C.H.; Gouker, F.; Crowell, C.R.; Evans, L.; DiFazio, S.P.; Smart, C.D.; Smart, L.B. Joint linkage and association mapping of complex traits in shrub willow (Salix purpurea L.). Ann. Bot. 2019, 124, 701–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mammadov, J.; Sun, X.; Gao, Y.; Ochsenfeld, C.; Bakker, E.; Ren, R.; Flora, J.; Wang, X.; Kumpatla, S.; Meyer, D.; et al. Combining powers of linkage and association mapping for precise dissection of QTL controlling resistance to gray leaf spot disease in maize (Zea mays L.). BMC Genom. 2015, 16, 916. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, G. Estimating the Dimension of a Model. Ann. Stat. 1978, 6, 461–464. [Google Scholar] [CrossRef]
- SAS Institute Inc. SAS® 9.4 Intelligence Platform: Data Administration Guide, 6th ed.; SAS Institute Inc.: Cary, NC, USA, 2016. [Google Scholar]
- Reif, J.C.; Zhao, Y.; Würschum, T.; Gowda, M.; Hahn, V. Genomic prediction of sunflower hybrid performance. Plant Breed. 2013, 132, 107–114. [Google Scholar] [CrossRef]
- Holm, S. A simple sequentially rejective multiple test procedure. Scand. J. Stat. 1979, 6, 65–70. [Google Scholar]
- Chen, J.; Zavala, C.; Ortega, N.; Petroli, C.; Franco, J.; Burgueño, J.; Costich, D.E.; Hearne, S.J. The Development of Quality Control Genotyping Approaches: A Case Study Using Elite Maize Lines. PLoS ONE 2016, 11, e0157236. [Google Scholar] [CrossRef]
- Barría, A.; Christensen, K.A.; Yoshida, G.M.; Correa, K.; Jedlicki, A.; Lhorente, J.P.; Davidson, W.S.; Yanez, J.M. Genomic Predictions and Genome-Wide Association Study of Resistance Against Piscirickettsia salmonis in Coho Salmon (Oncorhynchus kisutch) Using ddRAD Sequencing. G3: Genes|Genomes|Genetics 2018, 8, 1183–1194. [Google Scholar] [CrossRef] [Green Version]
- Gao, N.; Teng, J.; Ye, S.; Yuan, X.; Huang, S.; Zhang, H.; Zhang, X.; Li, J.; Zhang, Z. Genomic Prediction of Complex Phenotypes Using Genic Similarity Based Relatedness Matrix. Front. Genet. 2018, 9, 364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hochholdinger, F.; Hoecker, N. Towards the molecular basis of heterosis. Trends Plant Sci. 2007, 12, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Beyene, Y.; Gowda, M.; Suresh, L.M.; Mugo, S.; Olsen, M.; Oikeh, S.O.; Juma, C.; Tarekegne, A.; Prasanna, B.M. Genetic analysis of tropical maize inbred lines for resistance to maize lethal necrosis disease. Euphytica 2017, 213, 224. [Google Scholar] [CrossRef] [Green Version]
- Springer, N.M.; Stupar, R.M. Allelic variation and heterosis in maize: How do two halves make more than a whole? Genome Res. 2007, 17, 264–275. [Google Scholar] [CrossRef] [Green Version]
- Karume, K.; Nazir, N.; Salmin, I. Assessment of heterosis and combining ability in maize (Zea mays L.) for maize lethal necrosis disease. Afr. J. Plant Breed. 2017, 4, 104–200. [Google Scholar] [CrossRef] [Green Version]
- Mbega, E.; Ndakidemi, P.; Mamiro, D.; Mushongi, A.; Kitenge, K.; Ndomba, O. Role of Potyviruses in Synergistic Interaction Leading to Maize Lethal Necrotic Disease on Maize. Int. J. Curr. Microbiol. Appl. Sci. 2016, 5, 85–96. [Google Scholar] [CrossRef]
- Mackay, T.F.C. The genetic architecture of quantitative traits: Lessons from Drosophila, Current Opinion in Genetics and Development. Curr. Opin. Genet. Dev. 2004, 14, 253–257. [Google Scholar] [CrossRef]
- Forabosco, P.; Falchi, M.; Devoto, M. Statistical tools for linkage analysis and genetic association studies. Expert Rev. Mol. Diagn. 2005, 5, 781–796. [Google Scholar] [CrossRef]
- Gallois, J.-L.; Moury, B.; German-Retana, S. Role of the Genetic Background in Resistance to Plant Viruses. Int. J. Mol. Sci. 2018, 19, 2856. [Google Scholar] [CrossRef] [Green Version]
- Jansen, R.C.; Stam, P. High Resolution of Quantitative Traits into Multiple Loci via Interval Mapping. Genet. 1994, 136, 1447–1455. [Google Scholar]
- Jones, M.W.; Penning, B.W.; Jamann, T.M.; Glaubitz, J.C.; Romay, C.; Buckler, E.S.; Redinbaugh, M.G. Diverse chromosomal locations of quantitative trait loci for tolerance to maize chlorotic mottle virus in five maize populations. Phytopathology 2018, 108, 748–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gustafson, T.J.; de Leon, N.; Kaeppler, S.M.; Tracy, W.F. Genetic analysis of sugarcane mosaic virus resistance in the Wisconsin diversity panel of maize. Crop. Sci. 2018, 58, 1853–1865. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
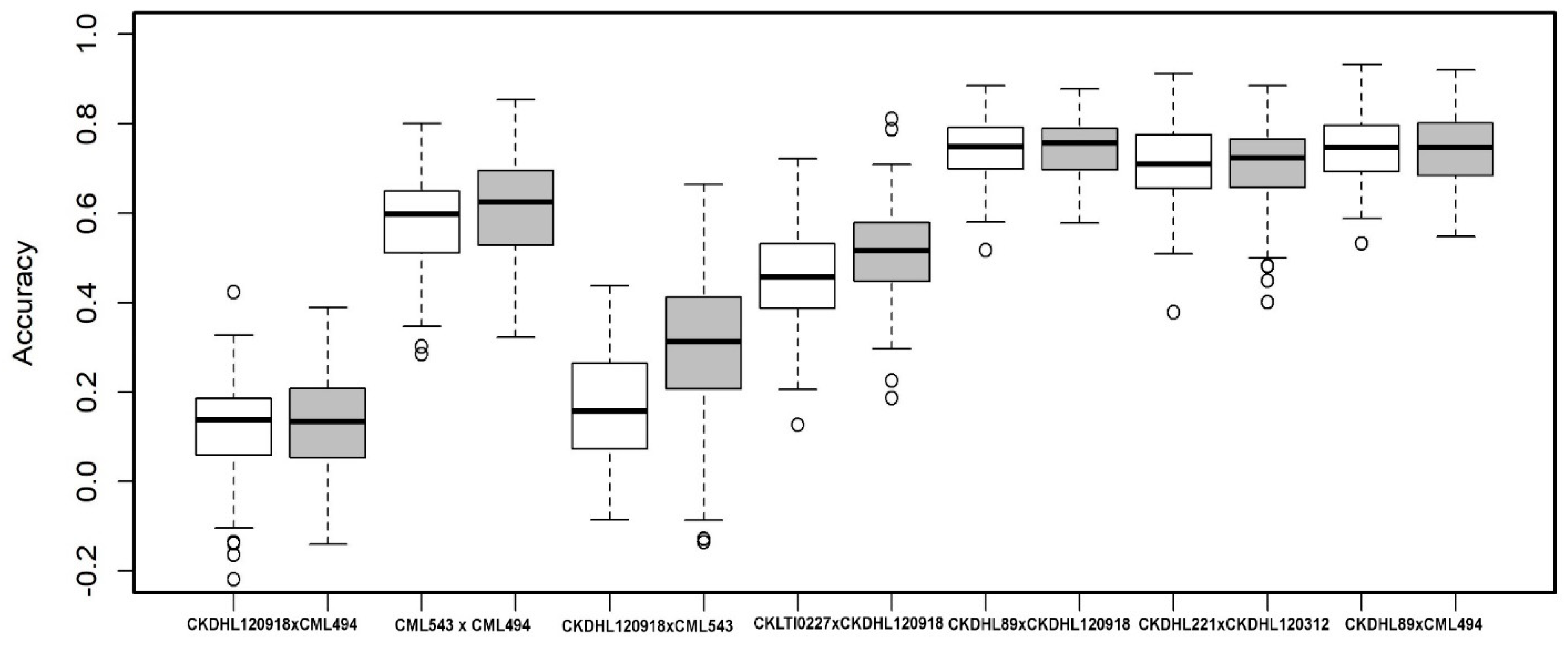{kind=link}
| Trait | Mean (Range) | σ2G | σ2e * | H2 |
|---|---|---|---|---|
| CKDHL120918 × CML494 (F3 pop1) | ||||
| MLN-DS | 4.52 (3.06–7.12) | 0.41 ** | 0.44 | 0.74 |
| AUDPC | 131.9 (92.9–185.8) | 272.20 ** | 230.73 | 0.78 |
| CML543 × CML494 (F3 pop2) | ||||
| MLN-DS | 5.11 (3.86–6.35) | 0.17 ** | 0.32 | 0.62 |
| AUDPC | 140.8 (103.8–168.9) | 141.05 ** | 177.68 | 0.70 |
| CKDHL120918 × CML543 (F3 pop3) | ||||
| MLN-DS | 4.52 (3.17–6.86) | 0.46 ** | 0.50 | 0.73 |
| AUDPC | 126.2 (82.1–201.0) | 282.50 ** | 280.87 | 0.75 |
| CKLTI0227 × CKDHL120918 (F3 pop4) | ||||
| MLN-DS | 4.60 (2.80–7.65) | 0.69 ** | 0.57 | 0.79 |
| AUDPC | 129.1 (92.6–194.7) | 467.78 ** | 299.86 | 0.82 |
| CKDHL0089 × CKDHL120918 (F3 pop5) | ||||
| MLN-DS | 4.94 (2.85–8.20) | 0.65 ** | 0.72 | 0.73 |
| AUDPC | 133.9 (79.9–209.3) | 411.12 ** | 379.39 | 0.76 |
| CKDHL0221 × CKDHL120312 (F3 pop6) | ||||
| MLN-DS | 4.40 (2.56–6.97) | 0.50 ** | 0.49 | 0.75 |
| AUDPC | 125.6 (78.0–187.9) | 374.25 ** | 245.35 | 0.82 |
| CKDHL0089 × CML494 (F3 pop7) | ||||
| MLN-DS | 4.82 (2.94–7.31) | 0.44 ** | 0.51 | 0.72 |
| AUDPC | 134.4 (91.0–191.8) | 300.75 ** | 260.51 | 0.78 |
| Across seven populations | ||||
| MLN-DS | 4.70 (2.56–8.20) | 0.40 ** | 0.50 | 0.70 |
| AUDPC | 132.4 (78.0–209.3) | 265.48 ** | 267.90 | 0.75 |
| Trait Name | QTL Name a | Chr | Position (cM) | LOD | PVE (%) | Add | Dom | Nature of QTL | Total PVE (%) | Marker Name | Physical Position (bp) | Fav Parent | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Left M | Right M | Left M | Right M | |||||||||||
| CKDHL120918 × CML494 | ||||||||||||||
| MLN-DS | qMLN1_265 | 1 | 183 | 3.99 | 2.58 | −0.46 | −0.26 | PD | 30.08 | PZB00648_5 | d8_3 | 17,595,139 | 265,199,938 | CKDHL120918 |
| qMLN2_156 | 2 | 3 | 4.13 | 2.34 | −0.04 | 1.09 | OD | PZA01232_1 | PHM3055_9 | 155,868,024 | 192,602,324 | CKDHL120918 | ||
| qMLN2_10 | 2 | 122 | 3.16 | 0.69 | 0.07 | −0.23 | OD | PZA00620_3 | PZA00365_2 | 10,429,405 | 1,221,385 | CML494 | ||
| qMLN3_142 | 3 | 23 | 23.92 | 4.27 | 0.37 | −0.21 | PD | PZA00920_1 | PHM15449_10 | 142,821,031 | 125,077,922 | CML494 | ||
| qMLN4_150 | 4 | 90 | 4.41 | 2.53 | −0.02 | 0.70 | OD | S4_153520131 | S4_149896839 | 153,520,131 | 149,896,839 | CKDHL120918 | ||
| qMLN5_177 | 5 | 88 | 4.24 | 2.58 | 0.03 | 0.83 | OD | PZA01410_1 | S5_177634071 | 172,682,963 | 177,634,071 | CML494 | ||
| qMLN6_85 | 6 | 78 | 14.11 | 3.98 | 0.34 | −0.20 | PD | PZB01009_1 | PHM8909_12 | 84,664,840 | 91,883,155 | CML494 | ||
| qMLN6_85 | 6 | 132 | 6.25 | 4.56 | 0.19 | 0.76 | OD | PZB01009_1 | PHM8909_12 | 84,664,840 | 918,83,155 | CML494 | ||
| qMLN7_158 | 7 | 87 | 4.71 | 2.48 | −0.07 | 1.11 | OD | PHM7898_10 | S7_157472460 | 161,993,743 | 157,472,460 | CKDHL120918 | ||
| qMLN9_142 | 9 | 66 | 3.64 | 1.02 | −0.11 | −0.21 | OD | PZB00221_3 | PHM229_15 | 142,271,047 | 30,003,189 | CKDHL120918 | ||
| qMLN10_9 | 10 | 4 | 3.82 | 2.43 | 0.08 | 0.84 | OD | PZA00866_2 | PHM5740_9 | 124,203,565 | 87,73,358 | CML494 | ||
| AUDPC | qMLN2_10 | 2 | 121 | 3.03 | 3.23 | 1.54 | −6.16 | OD | 34.72 | PZA00620_3 | PZA00365_2 | 10,429,405 | 1,221,385 | CML494 |
| qMLN3_142 | 3 | 23 | 12.68 | 8.92 | 5.50 | −5.57 | DO | PZA00920_1 | PHM15449_10 | 142,821,031 | 125,077,922 | CML494 | ||
| qMLN4_143 | 4 | 140 | 2.79 | 1.71 | −2.78 | 1.27 | PD | PHM1505_31 | S4_9850443 | 143,162,745 | 9,850,443 | CKDHL120918 | ||
| qMLN6_85 | 6 | 78 | 17.96 | 21.64 | 9.80 | −5.38 | PD | PZB01009_1 | PHM8909_12 | 84,664,840 | 91,883,155 | CML494 | ||
| qMLN9_142 | 9 | 68 | 4.79 | 6.25 | −4.08 | −5.16 | OD | PZB00221_3 | PHM229_15 | 142,271,047 | 30,003,189 | CKDHL120918 | ||
| CML543 × CML494 | ||||||||||||||
| MLN-DS | qMLN1_47 | 1 | 43 | 3.41 | 1.10 | −0.07 | 0.04 | PD | 43.49 | csu1138_4 | S1_46411896 | 119,018,556 | 46,411,896 | CML494 |
| qMLN3_146 | 3 | 14 | 11.79 | 4.08 | −0.72 | 0.25 | PD | S3_146966676 | S3_146363360 | 146,966,676 | 146,363,360 | CML494 | ||
| qMLN4_30 | 4 | 14 | 2.50 | 9.94 | −0.22 | 0.52 | OD | PZA02457_1 | bt2_7 | 29,031,200 | 66,290,994 | CML494 | ||
| qMLN5_160 | 5 | 3 | 3.52 | 1.22 | −0.08 | 0.04 | PD | S5_42297152 | PZA01796_1 | 42,297,152 | 160,321,846 | CML494 | ||
| qMLN6_21 | 6 | 4 | 5.03 | 1.64 | 0.09 | −0.05 | PD | S6_21007530 | PZA03063_21 | 21,007,530 | 25,335,225 | CML543 | ||
| qMLN8_10 | 8 | 84 | 3.42 | 12.37 | −0.21 | 0.53 | OD | S8_10001165 | PZA02388_1 | 10,001,165 | 169,137 | CML494 | ||
| qMLN9_142 | 9 | 3 | 31.21 | 12.72 | 0.70 | −0.08 | AD | PZA00832_1 | PHM7916_4 | 147,131,097 | 132,762,904 | CML543 | ||
| AUDPC | qMLN1_47 | 1 | 42 | 6.14 | 1.64 | −2.93 | 1.62 | PD | 49.84 | csu1138_4 | S1_46411896 | 119,018,556 | 46,411,896 | CML494 |
| qMLN1_265 | 1 | 154 | 2.97 | 4.17 | −7.58 | 9.26 | OD | d8_3 | umc128_2 | 265,199,938 | 227,602,208 | CML494 | ||
| qMLN3_146 | 3 | 14 | 15.65 | 4.18 | −24.01 | 7.57 | PD | S3_146966676 | S3_146363360 | 146,966,676 | 146,363,360 | CML494 | ||
| qMLN3_142 | 3 | 107 | 3.77 | 1.17 | 2.73 | 0.84 | PD | PZA00920_1 | PZA02402_1 | 142,821,031 | 169,771,952 | CML543 | ||
| qMLN4_30 | 4 | 14 | 3.88 | 7.99 | −6.91 | 15.27 | OD | PZA02457_1 | bt2_7 | 29,031,200 | 66,290,994 | CML494 | ||
| qMLN5_160 | 5 | 3 | 3.91 | 1.01 | −2.48 | 0.94 | PD | S5_42297152 | PZA01796_1 | 42,297,152 | 160,321,846 | CML494 | ||
| qMLN6_21 | 6 | 2 | 8.95 | 2.38 | 3.84 | −0.71 | AD | S6_21007530 | PZA03063_21 | 21,007,530 | 25,335,225 | CML543 | ||
| qMLN7_158 | 7 | 119 | 4.81 | 8.13 | 5.12 | −18.38 | OD | PHM7898_10 | PHM1912_23 | 161,993,743 | 155,970,264 | CML543 | ||
| qMLN8_10 | 8 | 85 | 3.05 | 5.28 | −6.73 | 9.00 | OD | S8_10001165 | PZA02388_1 | 10,001,165 | 169,137 | CML494 | ||
| qMLN9_142 | 9 | 3 | 34.18 | 10.54 | 21.95 | −4.51 | PD | PZA00832_1 | PHM7916_4 | 147,131,097 | 132,762,904 | CML543 | ||
| CKDHL120918 × CML543 | ||||||||||||||
| MLN-DS | qMLN1_265 | 1 | 14 | 4.12 | 2.79 | −0.04 | −0.21 | OD | 47.88 | d8_3 | PHM14475_7 | 265,199,938 | 256,245,118 | CKDHL120918 |
| qMLN1_252 | 1 | 28 | 3.31 | 3.08 | −0.12 | −0.15 | OD | PZA02269_4 | PHM4942_12 | 252,721,946 | 226,461,786 | CKDHL120918 | ||
| qMLN2_2 | 2 | 50 | 4.30 | 3.45 | −0.05 | −0.23 | OD | PHM13440_13 | PZA00365_2 | 2,527,344 | 1,221,385 | CKDHL120918 | ||
| qMLN3_142 | 3 | 73 | 33.33 | 28.84 | 0.46 | −0.26 | PD | S3_146966676 | S3_68596995 | 146,966,676 | 68,596,995 | CML543 | ||
| qMLN4_143 | 4 | 120 | 4.08 | 2.87 | 0.16 | −0.04 | PD | bt2_7 | PHM1505_31 | 66,290,994 | 143,162,745 | CML543 | ||
| qMLN5_202 | 5 | 112 | 2.89 | 4.43 | −0.14 | −0.19 | OD | PZB00765_1 | PHM5484_22 | 202,174,585 | 21,449,633 | CKDHL120918 | ||
| qMLN7_158 | 7 | 1 | 2.62 | 3.15 | −0.39 | −0.44 | DO | S7_157472460 | S7_137455469 | 157,472,460 | 137,455,469 | CKDHL120918 | ||
| qMLN10_9 | 10 | 81 | 6.77 | 5.10 | −0.15 | −0.22 | OD | PHM5740_9 | PZA01642_1 | 8,773,358 | 14,703,451 | CKDHL120918 | ||
| AUDPC | qMLN1_265 | 1 | 14 | 4.85 | 2.50 | −1.32 | −5.28 | OD | 52.80 | d8_3 | PHM14475_7 | 265,199,938 | 256,245,118 | CKDHL120918 |
| qMLN1_252 | 1 | 28 | 3.44 | 2.40 | −3.01 | −3.52 | DO | PZA02269_4 | PHM4942_12 | 252,721,946 | 226,461,786 | CKDHL120918 | ||
| qMLN2_2 | 2 | 50 | 4.03 | 2.46 | −0.78 | −5.45 | OD | PHM13440_13 | PZA00365_2 | 2,527,344 | 1,221,385 | CKDHL120918 | ||
| qMLN3_154 | 3 | 3 | 2.93 | 5.23 | 10.02 | −11.50 | DO | S3_154250438 | S3_150836832 | 154,250,438 | 150,836,832 | CML543 | ||
| qMLN3_142 | 3 | 74 | 17.68 | 11.09 | 7.15 | −6.24 | DO | S3_146966676 | S3_68596995 | 146,966,676 | 68,596,995 | CML543 | ||
| qMLN3_207 | 3 | 109 | 2.73 | 2.02 | −3.50 | −0.77 | PD | PZA00538_15 | PZA01931_17 | 206,889,707 | 227,682,081 | CKDHL120918 | ||
| qMLN4_143 | 4 | 120 | 6.03 | 3.30 | 4.58 | −0.86 | AD | bt2_7 | PHM1505_31 | 66,290,994 | 143,162,745 | CML543 | ||
| qMLN5_202 | 5 | 112 | 3.16 | 3.65 | −3.50 | −4.49 | OD | PZB00765_1 | PHM5484_22 | 202,174,585 | 21,449,633 | CKDHL120918 | ||
| qMLN6_157 | 6 | 118 | 3.83 | 4.79 | 11.14 | −8.68 | PD | PZA01618_2 | S6_156386857 | 129,927,781 | 156,386,857 | CML543 | ||
| qMLN7_158 | 7 | 1 | 3.85 | 3.30 | −10.90 | −11.74 | DO | S7_157472460 | S7_137455469 | 157,472,460 | 137,455,469 | CKDHL120918 | ||
| qMLN9_109 | 9 | 29 | 2.81 | 1.63 | −3.06 | −1.67 | PD | PHM229_15 | S9_109549230 | 30,003,189 | 109,549,230 | CKDHL120918 | ||
| qMLN10_41 | 10 | 2 | 2.90 | 3.46 | 9.86 | −10.22 | DO | PHM4066_11 | PHM1956_90 | 41,187,565 | 40,187,565 | CML543 | ||
| qMLN10_9 | 10 | 82 | 7.45 | 4.61 | −3.83 | −5.70 | OD | PHM5740_9 | PZA01642_1 | 8,773,358 | 14,703,451 | CKDHL120918 | ||
| CKLTI0227 × CKDHL120918 | ||||||||||||||
| MLN-DS | qMLN1_282 | 1 | 99 | 3.21 | 1.55 | 0.69 | −0.60 | DO | 25.13 | PHM4752_14 | PZA03020_8 | 298,874,066 | 282,044,048 | CKLTI0227 |
| qMLN1_265 | 1 | 109 | 3.52 | 1.56 | 0.68 | −0.63 | DO | PZA03020_8 | PZA01254_2 | 282,044,048 | 106,204,446 | CKLTI0227 | ||
| qMLN1_47 | 1 | 139 | 4.79 | 1.38 | 0.76 | −0.82 | DO | S1_22744948 | S1_87158930 | 22,744,948 | 87,158,930 | CKLTI0227 | ||
| qMLN1_27 | 1 | 202 | 3.54 | 0.91 | 0.93 | −0.94 | DO | S1_15353866 | S1_2693968 | 15,353,866 | 2,693,968 | CKLTI0227 | ||
| qMLN3_142 | 3 | 37 | 4.44 | 1.45 | 0.73 | −0.35 | PD | S3_146966676 | S3_55444954 | 146,966,676 | 55,444,954 | CKLTI0227 | ||
| qMLN3_130 | 3 | 65 | 5.68 | 1.48 | 0.70 | −0.82 | DO | S3_92864540 | S3_136165565 | 92,864,540 | 136,165,565 | CKLTI0227 | ||
| qMLN3_130 | 3 | 121 | 5.32 | 1.49 | −0.68 | −1.08 | OD | PHM2343_25 | S3_154250438 | 27,981,649 | 154,250,438 | CKDHL120918 | ||
| qMLN4_30 | 4 | 36 | 4.36 | 0.35 | 0.72 | −0.05 | AD | PZA02457_1 | PZA00726_10 | 29,031,200 | 60,768,063 | CKLTI0227 | ||
| qMLN4_150 | 4 | 96 | 4.29 | 1.59 | 0.59 | −0.74 | OD | S4_155296684 | S4_9741874 | 155,296,684 | 9,741,874 | CKLTI0227 | ||
| qMLN5_160 | 5 | 130 | 4.12 | 1.05 | 0.90 | −0.83 | DO | S5_154350617 | S5_198716574 | 154,350,617 | 198,716,574 | CKLTI0227 | ||
| qMLN5_42 | 5 | 161 | 3.49 | 0.25 | −0.08 | −1.02 | OD | S5_42297152 | PZA02164_16 | 42,297,152 | 112,179,855 | CKDHL120918 | ||
| qMLN6_157 | 6 | 20 | 5.87 | 1.46 | 0.59 | −1.12 | OD | S6_156386857 | PHM4748_16 | 156,386,857 | 158,540,019 | CKLTI0227 | ||
| qMLN6_85 | 6 | 29 | 7.17 | 1.68 | 0.72 | −0.34 | PD | PHM4748_16 | PZB01009_1 | 158,540,019 | 84,664,840 | CKLTI0227 | ||
| qMLN6_85 | 6 | 83 | 8.86 | 1.79 | 0.81 | −0.43 | PD | PZB01009_1 | S6_89823772 | 84,664,840 | 89,823,772 | CKLTI0227 | ||
| qMLN6_157 | 6 | 116 | 4.73 | 1.10 | 0.07 | 1.37 | OD | PHM2658_129 | PZA01884_1 | 164,999,578 | 132,316,835 | CKLTI0227 | ||
| qMLN7_158 | 7 | 145 | 3.56 | 1.08 | 0.86 | −0.99 | DO | S7_167230991 | S7_127970539 | 167,230,991 | 127,970,539 | CKLTI0227 | ||
| qMLN8_10 | 8 | 100 | 4.34 | 1.37 | 0.73 | −0.90 | OD | PZA02746_2 | PZA02388_1 | 163,067,200 | 169,137 | CKLTI0227 | ||
| qMLN9_109 | 9 | 9 | 3.19 | 1.64 | −0.63 | −0.57 | DO | PZA00708_3 | S9_109549230 | 147,381,231 | 109,549,230 | CKDHL120918 | ||
| qMLN10_114 | 10 | 36 | 5.74 | 1.61 | 0.69 | −0.83 | OD | S10_113832226 | PZA01001_2 | 113,832,226 | 146,538,889 | CKLTI0227 | ||
| AUDPC | qMLN1_265 | 1 | 163 | 6.85 | 1.35 | 24.93 | −8.24 | PD | 28.69 | d8_3 | PZA02269_4 | 265,199,938 | 252,721,946 | CKLTI0227 |
| qMLN2_41 | 2 | 2 | 12.20 | 2.31 | 40.41 | −11.93 | PD | PHM10404_8 | PZA02450_1 | 40,967,991 | 47,575,949 | CKLTI0227 | ||
| qMLN3_142 | 3 | 34 | 5.67 | 3.42 | 14.61 | −8.98 | PD | S3_146966676 | S3_55444954 | 146,966,676 | 55,444,954 | CKLTI0227 | ||
| qMLN3_146 | 3 | 113 | 10.79 | 1.98 | −40.42 | −5.50 | AD | S3_146250249 | S3_146026612 | 146,250,249 | 146,026,612 | CKDHL120918 | ||
| qMLN3_130 | 3 | 125 | 3.49 | 3.81 | 12.36 | −18.09 | OD | PHM2343_25 | S3_154250438 | 27,981,649 | 154,250,438 | CKLTI0227 | ||
| qMLN4_30 | 4 | 77 | 3.29 | 2.44 | 19.01 | −20.07 | DO | S4_6601124 | PZA02509_15 | 6,601,124 | 3,904,858 | CKLTI0227 | ||
| qMLN6_157 | 6 | 32 | 8.84 | 3.77 | 16.09 | −5.31 | PD | PHM4748_16 | PZB01009_1 | 158,540,019 | 84,664,840 | CKLTI0227 | ||
| qMLN9_147 | 9 | 6 | 3.28 | 3.17 | −15.11 | −9.92 | PD | S9_146012201 | PZA00708_3 | 146,012,201 | 147,381,231 | CKDHL120918 | ||
| qMLN9_109 | 9 | 21 | 3.19 | 2.66 | 18.29 | −19.22 | DO | PZA00708_3 | S9_109549230 | 147,381,231 | 109,549,230 | CKLTI0227 | ||
| qMLN10_114 | 10 | 36 | 3.54 | 3.20 | 16.73 | −18.91 | DO | S10_113832226 | PZA01001_2 | 113,832,226 | 146,538,889 | CKLTI0227 | ||
| CKDHL0089 × CKDHL120918 | ||||||||||||||
| MLN-DS | qMLN1_18 | 1 | 4 | 4.42 | 7.15 | −0.11 | 0.87 | OD | 54.34 | S1_18838432 | PZA00175_2 | 18,838,432 | 8,510,027 | CKDHL120918 |
| qMLN2_169 | 2 | 168 | 4.25 | 6.61 | −0.06 | 1.07 | OD | PZA02727_1 | PZA00515_10 | 227,921,381 | 169,265,278 | CKDHL120918 | ||
| qMLN3_142 | 3 | 73 | 44.14 | 24.44 | 0.62 | −0.10 | AD | PZA00920_1 | S3_133048570 | 142,821,031 | 133,048,570 | CKDHL0089 | ||
| qMLN5_190 | 5 | 128 | 3.75 | 1.56 | −0.14 | −0.06 | PD | S5_190675983 | S5_201226926 | 190,675,983 | 201,226,926 | CKDHL120918 | ||
| qMLN6_157 | 6 | 20 | 3.21 | 8.20 | 0.31 | −0.41 | OD | S6_156386857 | PHM3466_69 | 156,386,857 | 167,148,384 | CKDHL0089 | ||
| qMLN6_85 | 6 | 126 | 4.16 | 1.78 | 0.16 | 0.04 | PD | PZA00942_2 | PHM8909_12 | 102,566,000 | 91,883,155 | CKDHL0089 | ||
| qMLN10_9 | 10 | 4 | 12.06 | 6.32 | −0.28 | −0.10 | PD | PZA01313_2 | PHM5740_9 | 3,598,262 | 8,773,358 | CKDHL120918 | ||
| AUDPC | qMLN1_18 | 1 | 10 | 3.69 | 8.65 | −3.67 | 15.28 | OD | 57.62 | S1_18838432 | PZA00175_2 | 18,838,432 | 8,510,027 | CKDHL120918 |
| qMLN2_169 | 2 | 166 | 3.18 | 6.28 | −1.81 | 21.55 | OD | PZA02727_1 | PZA00515_10 | 227,921,381 | 169,265,278 | CKDHL120918 | ||
| qMLN3_142 | 3 | 73 | 50.02 | 27.46 | 16.42 | −2.21 | AD | PZA00920_1 | S3_133048570 | 142,821,031 | 133,048,570 | CKDHL0089 | ||
| qMLN5_190 | 5 | 127 | 5.49 | 2.11 | −4.27 | −1.58 | PD | PHM7908_25 | S5_190675983 | 191,075,472 | 190,675,983 | CKDHL120918 | ||
| qMLN6_157 | 6 | 21 | 2.96 | 8.12 | 7.34 | −10.37 | OD | S6_156386857 | PHM3466_69 | 156,386,857 | 167,148,384 | CKDHL0089 | ||
| qMLN6_85 | 6 | 133 | 5.03 | 1.96 | 4.23 | 0.36 | AD | PHM8909_12 | PZA00427_3 | 91,883,155 | 79,815,961 | CKDHL0089 | ||
| qMLN10_9 | 10 | 3 | 11.23 | 5.20 | −6.46 | −2.44 | PD | PZA01313_2 | PHM5740_9 | 3,598,262 | 8,773,358 | CKDHL120918 | ||
| CKDHL0221 × CKDHL120312 | ||||||||||||||
| MLN-DS | qMLN1_47 | 1 | 71 | 5.32 | 3.74 | −0.16 | 0.07 | PD | 52.13 | PZA00447_8 | S1_46411896 | 9,024,005 | 46,411,896 | CKDHL120312 |
| qMLN3_130 | 3 | 56 | 44.83 | 44.51 | 0.56 | −0.11 | AD | PZA02402_1 | PHM15449_10 | 169,771,952 | 125,077,922 | CKDHL0221 | ||
| qMLN4_150 | 4 | 37 | 2.66 | 2.74 | −0.13 | −0.09 | PD | PZA01187_1 | PHM1505_31 | 177,666,738 | 143,162,745 | CKDHL120312 | ||
| qMLN4_7 | 4 | 128 | 4.63 | 3.23 | 0.15 | 0.01 | AD | S4_6544767 | PHM16788_6 | 6,544,767 | 13,581,955 | CKDHL0221 | ||
| qMLN8_10 | 8 | 45 | 4.26 | 3.12 | 0.15 | 0.03 | AD | PHM5235_8 | PZA00368_1 | 94,414,978 | 5,632,308 | CKDHL0221 | ||
| AUDPC | qMLN1_47 | 1 | 71 | 4.97 | 4.38 | −4.05 | 1.41 | PD | 59.07 | PZA00447_8 | S1_46411896 | 9,024,005 | 46,411,896 | CKDHL120312 |
| qMLN1_47 | 1 | 100 | 2.98 | 2.49 | 2.21 | −2.90 | OD | S1_46411896 | PHM12323_17 | 46,411,896 | 53,357,797 | CKDHL0221 | ||
| qMLN3_130 | 3 | 56 | 24.64 | 26.00 | 9.70 | −2.76 | PD | PZA02402_1 | PHM15449_10 | 169,771,952 | 125,077,922 | CKDHL0221 | ||
| qMLN3_142 | 3 | 63 | 13.19 | 12.71 | 6.76 | −1.85 | PD | PZA00279_2 | PZA00920_1 | 52,804,070 | 142,821,031 | CKDHL0221 | ||
| qMLN4_150 | 4 | 39 | 3.65 | 5.24 | −4.27 | −2.01 | PD | PZA01187_1 | PHM1505_31 | 177,666,738 | 143,162,745 | CKDHL120312 | ||
| qMLN4_7 | 4 | 128 | 3.95 | 3.44 | 3.71 | 0.18 | AD | S4_6544767 | PHM16788_6 | 6,544,767 | 13,581,955 | CKDHL0221 | ||
| qMLN5_42 | 5 | 34 | 2.63 | 2.36 | −0.23 | −4.17 | OD | PHM16854_3 | PZA00522_12 | 34,587,029 | 57,933,548 | CKDHL120312 | ||
| qMLN8_10 | 8 | 45 | 5.06 | 4.70 | 4.16 | −0.42 | AD | PHM5235_8 | PZA00368_1 | 94,414,978 | 5,632,308 | CKDHL0221 | ||
| qMLN10_114 | 10 | 43 | 5.16 | 4.47 | 3.72 | −2.52 | PD | PZA00814_1 | PHM1576_25 | 87,194,491 | 124,203,168 | CKDHL0221 | ||
| CKDHL0089 × CML494 | ||||||||||||||
| MLN-DS | qMLN5_190 | 5 | 89 | 4.40 | 5.48 | −0.17 | 0.03 | AD | 46.74 | PZA01427_1 | PHM7908_25 | 23,135,578 | 191,075,472 | CML494 |
| qMLN5_202 | 5 | 111 | 8.94 | 7.97 | −0.20 | −0.03 | AD | S5_200938637 | PHM563_9 | 200,938,637 | 204,993,639 | CML494 | ||
| qMLN6_157 | 6 | 107 | 3.37 | 8.42 | 0.09 | 0.71 | OD | S6_157568432 | S6_156386857 | 157,568,432 | 156,386,857 | CKDHL0089 | ||
| AUDPC | qMLN3_130 | 3 | 1 | 3.02 | 6.64 | 0.75 | −4.18 | OD | 50.87 | PZA01447_1 | S3_133048570 | 53,549,251 | 133,048,570 | CKDHL0089 |
| qMLN5_190 | 5 | 89 | 5.14 | 15.91 | −4.78 | 0.81 | AD | PZA01427_1 | PHM7908_25 | 23,135,578 | 191,075,472 | CML494 | ||
| qMLN5_202 | 5 | 111 | 7.62 | 16.65 | −4.81 | −0.76 | AD | S5_200938637 | PHM563_9 | 20,0938,637 | 204,993,639 | CML494 | ||
| qMLN6_157 | 6 | 108 | 3.20 | 14.95 | 2.34 | 16.81 | OD | S6_157568432 | S6_156386857 | 157,568,432 | 156,386,857 | CKDHL0089 | ||
| Marker | QTL Name | Chr | Position (Mbp) | Model A | Model B | Model C | |||
|---|---|---|---|---|---|---|---|---|---|
| α-Effect | PVE (%) | α-Effect | PVE (%) | α-Effect | PVE (%) | ||||
| PZA00447_8 | qMLN1_9 | 1 | 9.02 | −0.08 | 0.60 | – | – | – | – |
| PHM5622_21 | qMLN1_184 | 1 | 183.83 | – | – | – | – | −0.12 | 0.40 |
| S3_48493677 | qMLN3_48 | 3 | 48.49 | 0.32 | 7.90 | – | – | −0.02 | 0.40 |
| S3_55444954 | qMLN3_55 | 3 | 55.44 | 0.14 | 0.10 | 0.10 | 1.00 | – | – |
| S3_68596995 | qMLN3_68 | 3 | 68.60 | – | – | −0.06 | 0.20 | 0.06 | 0.40 |
| S3_92694873 | qMLN3_92 | 3 | 92.69 | −0.28 | 0.70 | – | – | −0.02 | 1.80 |
| S3_113429913 | qMLN3_113 | 3 | 113.43 | – | – | −0.38 | 1.00 | – | – |
| PHM15449_10 | qMLN3_125 | 3 | 125.08 | 0.10 | 3.30 | 0.11 | 0.80 | 0.16 | 2.20 |
| S3_148291047 | qMLN3_148 | 3 | 148.29 | −0.72 | 10.20 | −0.66 | 4.70 | −0.16 | 1.40 |
| S3_151342843 | qMLN3_151 | 3 | 151.34 | −0.23 | 3.20 | −0.42 | 3.30 | – | – |
| PHM2919_23 | qMLN3_199 | 3 | 199.89 | −0.12 | 0.40 | −0.12 | 0.40 | – | – |
| PZA00726_8 | qMLN4_60 | 4 | 60.77 | – | – | – | – | −0.04 | 0.70 |
| S4_235381719 | qMLN4_235 | 4 | 235.38 | – | – | – | – | 0.03 | 0.90 |
| PHM565_31 | qMLN5_24 | 5 | 24.24 | – | – | −0.03 | 0.00 | −0.47 | 0.30 |
| S5_170023563 | qMLN5_170 | 5 | 170.02 | – | – | – | – | 0.01 | 0.10 |
| PHM7908_25 | qMLN5_191 | 5 | 191.08 | – | – | – | – | 0.04 | 0.20 |
| S5_196017729 | qMLN5_196 | 5 | 196.02 | −0.11 | 0.10 | −0.03 | 0.10 | −0.01 | 0.20 |
| S5_202816906 | qMLN5_202 | 5 | 202.82 | – | – | – | – | −0.10 | 0.10 |
| PHM563_9 | qMLN5_204 | 5 | 204.99 | – | – | – | – | −0.08 | 0.20 |
| PZA03167_5 | qMLN5_207 | 5 | 207.60 | – | – | – | – | 0.32 | 0.30 |
| S5_209467974 | qMLN5_209 | 5 | 209.47 | – | – | – | – | −0.07 | 0.30 |
| S6_13300385 | qMLN6_13 | 6 | 13.30 | 0.19 | 1.90 | 0.20 | 1.10 | – | – |
| S6_86475982 | qMLN6_86 | 6 | 86.48 | −0.27 | 1.00 | – | – | – | – |
| S6_89823772 | qMLN6_90 | 6 | 89.82 | −0.24 | 2.70 | −0.23 | 0.90 | −0.22 | 2.50 |
| PHM5235_8 | qMLN8_94 | 8 | 94.41 | 0.15 | 0.50 | – | – | – | – |
| PZA01313_2 | qMLN10_4 | 10 | 3.60 | 0.11 | 1.30 | 0.18 | 2.90 | 0.10 | 0.80 |
| PHM5740_9 | qMLN10_9 | 10 | 8.77 | 0.09 | 0.50 | – | – | – | – |
| Total PVE (%) | 34.40 | 27.30 | 29.10 | ||||||
| PZA00447_8 | qMLN1_9 | 1 | 9.02 | −1.63 | 0.20 | – | – | – | – |
| PHM5622_21 | qMLN1_184 | 1 | 183.83 | – | – | – | – | −3.54 | 0.40 |
| S3_48493677 | qMLN3_48 | 3 | 48.49 | 6.63 | 6.80 | – | – | – | – |
| S3_55444954 | qMLN3_55 | 3 | 55.44 | 3.53 | 1.00 | 1.94 | 0.60 | – | – |
| S3_68596995 | qMLN3_68 | 3 | 68.60 | – | – | −2.45 | 0.60 | −1.55 | 2.30 |
| PHM15449_10 | qMLN3_125 | 3 | 125.08 | – | – | 3.01 | 1.00 | 3.65 | 3.00 |
| S3_148291047 | qMLN3_148 | 3 | 148.29 | −20.14 | 10.20 | −16.47 | 4.80 | −3.86 | 1.50 |
| S3_151342843 | qMLN3_151 | 3 | 151.34 | −12.06 | 4.90 | −10.59 | 3.70 | −4.72 | 1.80 |
| PHM2919_23 | qMLN3_199 | 3 | 199.89 | −4.35 | 0.50 | −4.04 | 0.80 | −1.70 | 0.80 |
| PZA00726_8 | qMLN4_60 | 4 | 60.77 | – | – | – | – | −1.64 | 0.70 |
| S4_155378923 | qMLN4_155 | 4 | 155.38 | – | – | −8.03 | 1.00 | – | – |
| S4_235381719 | qMLN4_235 | 4 | 235.38 | – | – | – | – | −10.74 | 0.80 |
| S5_170164477 | qMLN5_170 | 5 | 170.16 | 5.53 | 0.50 | – | – | 10.13 | 0.50 |
| PHM7908_25 | qMLN5_191 | 5 | 191.08 | – | – | – | – | 1.38 | 0.20 |
| S5_196017729 | qMLN5_196 | 5 | 196.02 | −2.14 | 0.20 | −1.13 | 0.40 | −4.37 | 0.20 |
| S5_202816906 | qMLN5_202 | 5 | 202.82 | – | – | – | – | –2.53 | 0.10 |
| PHM563_9 | qMLN5_204 | 5 | 204.99 | – | – | – | – | –5.61 | 0.30 |
| PZA03167_5 | qMLN5_207 | 5 | 207.60 | – | – | – | – | 7.57 | 0.30 |
| S5_209467974 | qMLN5_209 | 5 | 209.47 | – | – | – | – | 2.00 | 0.40 |
| S6_13300385 | qMLN6_13 | 6 | 13.30 | 6.30 | 4.30 | 5.69 | 1.40 | 0.54 | 1.00 |
| S6_86475982 | qMLN6_86 | 6 | 86.48 | –6.97 | 0.90 | – | – | – | – |
| S6_89823772 | qMLN6_90 | 6 | 89.82 | –6.58 | 0.90 | –6.14 | 1.00 | –5.81 | 2.00 |
| S8_74144408 | qMLN8_74 | 8 | 74.14 | – | – | – | – | 3.86 | 0.30 |
| PHM5235_8 | qMLN8_94 | 8 | 94.41 | 4.41 | 1.50 | – | – | – | – |
| S8_102533570 | qMLN8_102 | 8 | 102.53 | – | – | – | – | 0.83 | 0.30 |
| PZA01313_2 | qMLN10_4 | 10 | 3.60 | 3.41 | 1.20 | 4.73 | 3.30 | 0.66 | 0.90 |
| PHM5740_9 | qMLN10_9 | 10 | 8.77 | – | – | – | – | –5.51 | 0.30 |
| PZA00866_2 | qMLN10_124 | 10 | 124.20 | 1.71 | 0.60 | 1.44 | 0.40 | 1.79 | 0.90 |
| Total PVE (%) | 33.60 | 29.00 | 39.80 | ||||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Awata, L.A.O.; Beyene, Y.; Gowda, M.; L. M., S.; Jumbo, M.B.; Tongoona, P.; Danquah, E.; Ifie, B.E.; Marchelo-Dragga, P.W.; Olsen, M.; et al. Genetic Analysis of QTL for Resistance to Maize Lethal Necrosis in Multiple Mapping Populations. Genes 2020, 11, 32. https://doi.org/10.3390/genes11010032
Awata LAO, Beyene Y, Gowda M, L. M. S, Jumbo MB, Tongoona P, Danquah E, Ifie BE, Marchelo-Dragga PW, Olsen M, et al. Genetic Analysis of QTL for Resistance to Maize Lethal Necrosis in Multiple Mapping Populations. Genes. 2020; 11(1):32. https://doi.org/10.3390/genes11010032
Chicago/Turabian StyleAwata, Luka A. O., Yoseph Beyene, Manje Gowda, Suresh L. M., McDonald B. Jumbo, Pangirayi Tongoona, Eric Danquah, Beatrice E. Ifie, Philip W. Marchelo-Dragga, Michael Olsen, and et al. 2020. "Genetic Analysis of QTL for Resistance to Maize Lethal Necrosis in Multiple Mapping Populations" Genes 11, no. 1: 32. https://doi.org/10.3390/genes11010032
APA StyleAwata, L. A. O., Beyene, Y., Gowda, M., L. M., S., Jumbo, M. B., Tongoona, P., Danquah, E., Ifie, B. E., Marchelo-Dragga, P. W., Olsen, M., Ogugo, V., Mugo, S., & Prasanna, B. M. (2020). Genetic Analysis of QTL for Resistance to Maize Lethal Necrosis in Multiple Mapping Populations. Genes, 11(1), 32. https://doi.org/10.3390/genes11010032