The Many Faces of DFNB9: Relating OTOF Variants to Hearing Impairment


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
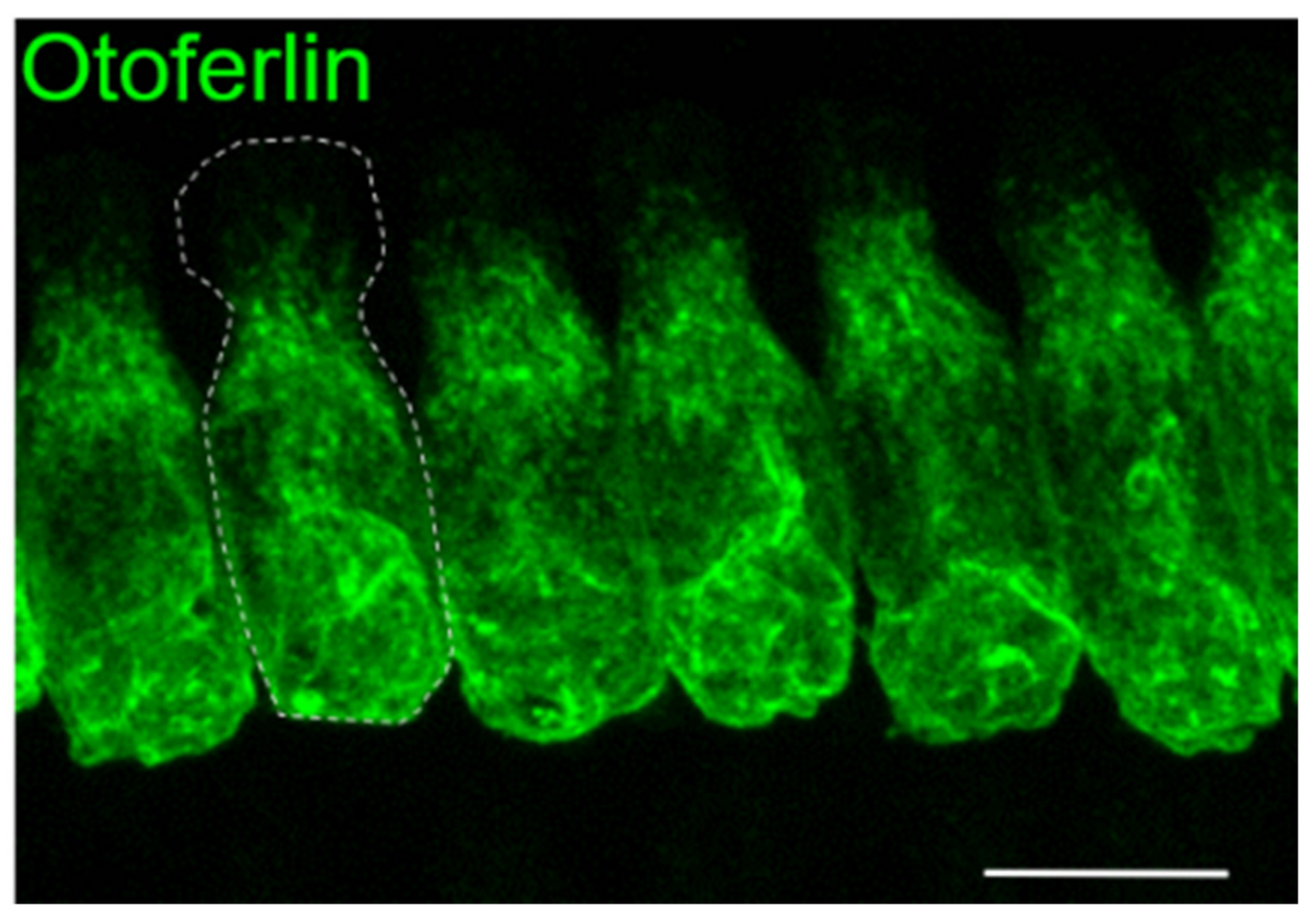
1.1. Mouse Studies Reveal Insights into Otoferlin Function
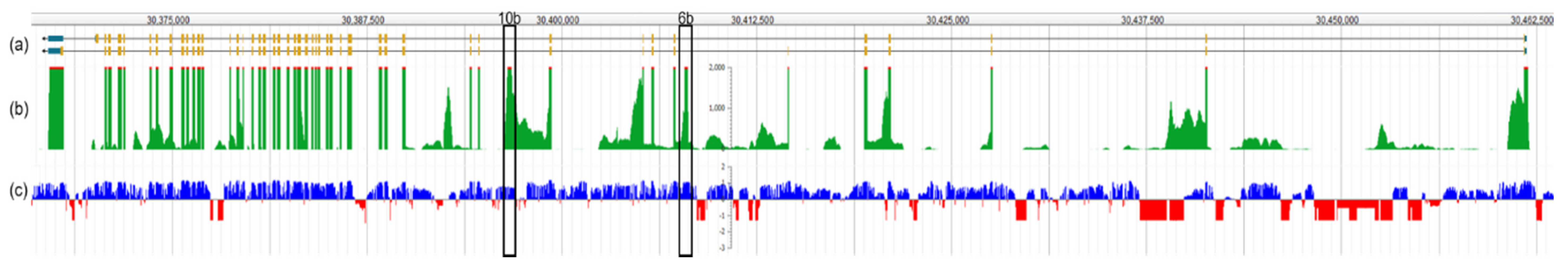
1.2. Otoferlin Isoforms
2. Hallmarks of Audiometric Testing in DFNB9 Patients
3. Molecular Epidemiology of OTOF-Associated Hearing Loss
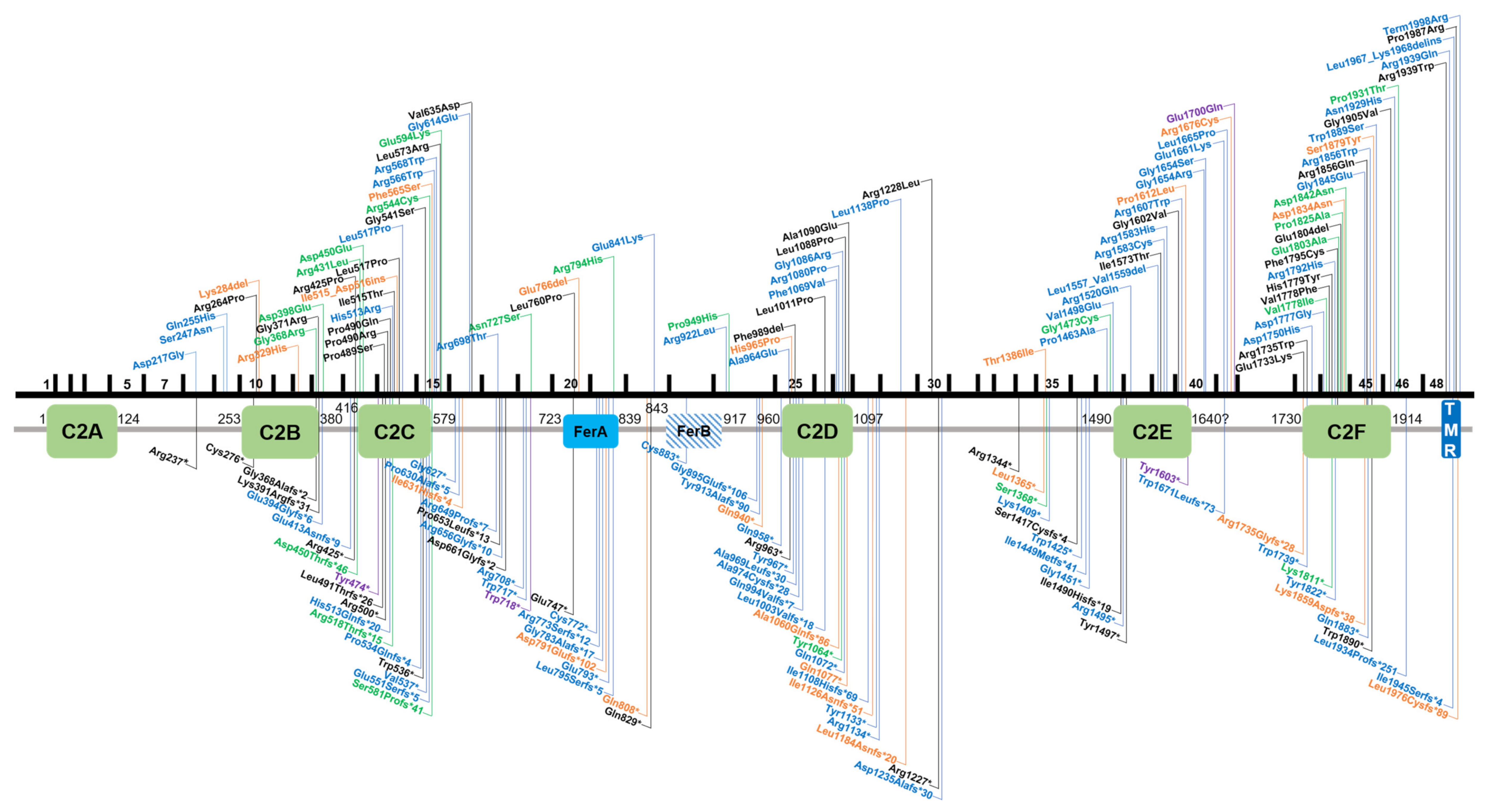
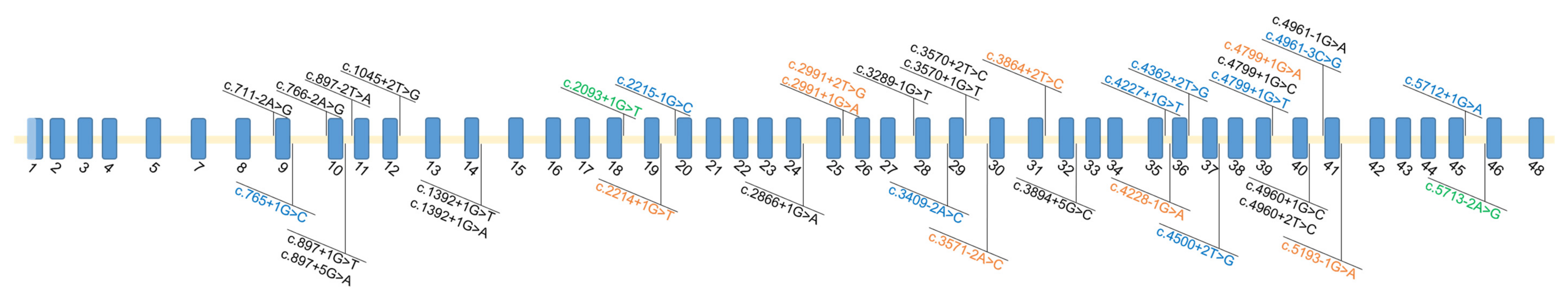
3.1. Summary of Variants Identified in Otoferlin
3.2. Population-Based Diagnostic Rates of Otoferlin
3.3. Diagnostic Rates of Otoferlin in Patients with Auditory Neuropathy/Synaptopathy
3.4. Missing Variants
4. Genotype-Phenotype Correlations in DFNB9 Patients
4.1. Temperature-Sensitive Auditory Synaptopathy
4.2. Progressive Hearing Impairment
5. Localization and Presumed Effects of Single Amino Acid Substitutions in Otoferlin
6. Current and Future Therapies for DFNB9
7. Outlook and Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Morton, C.C.; Nance, W.E. Newborn Hearing Screening—A Silent Revolution. N. Engl. J. Med. 2006, 354, 2151–2164. [Google Scholar] [CrossRef] [PubMed]
- Azaiez, H.; Booth, K.T.; Ephraim, S.S.; Crone, B.; Black-Ziegelbein, E.A.; Marini, R.J.; Shearer, A.E.; Sloan-Heggen, C.M.; Kolbe, D.; Casavant, T.; et al. Genomic Landscape and Mutational Signatures of Deafness-Associated Genes. Am. J. Hum. Genet. 2018, 103, 484–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaïb, H.; Place, C.; Salem, N.; Chardenoux, S.; Vincent, C.; Weissenbach, J.; El-Zir, E.; Loiselet, J.; Petit, C. A gene responsible for a sensorineural nonsyndromic recessive deafness maps to chromosome 2p22-23. Hum. Mol. Genet. 1996, 5, 155–158. [Google Scholar] [CrossRef] [Green Version]
- Yasunaga, S.; Grati, M.; Cohen-Salmon, M.; El-Amraoui, A.; Mustapha, M.; Salem, N.; El-Zir, E.; Loiselet, J.; Petit, C. A mutation in OTOF, encoding otoferlin, a FER-1-like protein, causes DFNB9, a nonsyndromic form of deafness. Nat. Genet. 1999, 21, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Roux, I.; Safieddine, S.; Nouvian, R.; Grati, M.; Simmler, M.-C.; Bahloul, A.; Perfettini, I.; Le Gall, M.; Rostaing, P.; Hamard, G.; et al. Otoferlin, defective in a human deafness form, is essential for exocytosis at the auditory ribbon synapse. Cell 2006, 127, 277–289. [Google Scholar] [CrossRef] [PubMed]
- Dulon, D.; Safieddine, S.; Jones, S.M.; Petit, C. Otoferlin Is Critical for a Highly Sensitive and Linear Calcium-Dependent Exocytosis at Vestibular Hair Cell Ribbon Synapses. J. Neurosci. 2009, 29, 10474–10487. [Google Scholar] [CrossRef] [Green Version]
- Beurg, M.; Safieddine, S.; Roux, I.; Bouleau, Y.; Petit, C.; Dulon, D. Calcium- and otoferlin-dependent exocytosis by immature outer hair cells. J. Neurosci. Off. J. Soc. Neurosci. 2008, 28, 1798–1803. [Google Scholar] [CrossRef] [Green Version]
- Pangrsic, T.; Lasarow, L.; Reuter, K.; Takago, H.; Schwander, M.; Riedel, D.; Frank, T.; Tarantino, L.M.; Bailey, J.S.; Strenzke, N.; et al. Hearing requires otoferlin-dependent efficient replenishment of synaptic vesicles in hair cells. Nat. Neurosci. 2010, 13, 869–876. [Google Scholar] [CrossRef]
- Beurg, M.; Michalski, N.; Safieddine, S.; Bouleau, Y.; Schneggenburger, R.; Chapman, E.R.; Petit, C.; Dulon, D. Control of exocytosis by synaptotagmins and otoferlin in auditory hair cells. J. Neurosci. Off. J. Soc. Neurosci. 2010, 30, 13281–13290. [Google Scholar] [CrossRef] [Green Version]
- Nouvian, R.; Neef, J.; Bulankina, A.V.; Reisinger, E.; Pangršič, T.; Frank, T.; Sikorra, S.; Brose, N.; Binz, T.; Moser, T. Exocytosis at the hair cell ribbon synapse apparently operates without neuronal SNARE proteins. Nat. Neurosci. 2011, 14, 411–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pangršič, T.; Reisinger, E.; Moser, T. Otoferlin: A multi-C2 domain protein essential for hearing. Trends Neurosci. 2012, 35, 671–680. [Google Scholar] [CrossRef] [PubMed]
- Michalski, N.A.; Goutman, J.D.; Auclair, S.M.; De Monvel, J.B.; Tertrais, M.; Emptoz, A.; Parrin, A.; Nouaille, S.; Guillon, M.; Sachse, M.; et al. Otoferlin acts as a Ca2+ sensor for vesicle fusion and vesicle pool replenishment at auditory hair cell ribbon synapses. eLife 2017, 6, e31013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strenzke, N.; Chakrabarti, R.; Al-Moyed, H.; Müller, A.; Hoch, G.; Pangrsic, T.; Yamanbaeva, G.; Lenz, C.; Pan, K.-T.; Auge, E.; et al. Hair cell synaptic dysfunction, auditory fatigue and thermal sensitivity in otoferlin Ile515Thr mutants. EMBO J. 2016, 35, e201694564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwander, M.; Sczaniecka, A.; Grillet, N.; Bailey, J.S.; Avenarius, M.; Najmabadi, H.; Steffy, B.M.; Federe, G.C.; Lagler, E.A.; Banan, R.; et al. A Forward Genetics Screen in Mice Identifies Recessive Deafness Traits and Reveals That Pejvakin Is Essential for Outer Hair Cell Function. J. Neurosci. 2007, 27, 2163–2175. [Google Scholar] [CrossRef] [Green Version]
- Duncker, S.V.; Franz, C.; Kuhn, S.; Schulte, U.; Campanelli, D.; Brandt, N.; Hirt, B.; Fakler, B.; Blin, N.; Ruth, P.; et al. Otoferlin Couples to Clathrin-Mediated Endocytosis in Mature Cochlear Inner Hair Cells. J. Neurosci. 2013, 33, 9508–9519. [Google Scholar] [CrossRef] [Green Version]
- Jung, S.; Maritzen, T.; Wichmann, C.; Jing, Z.; Neef, A.; Revelo, N.H.; Al-Moyed, H.; Meese, S.; Wojcik, S.M.; Panou, I.; et al. Disruption of adaptor protein 2μ (AP-2μ) in cochlear hair cells impairs vesicle reloading of synaptic release sites and hearing. EMBO J. 2015, 34, 2686–2702. [Google Scholar] [CrossRef]
- Yasunaga, S.; Grati, M.; Chardenoux, S.; Smith, T.N.; Friedman, T.B.; Lalwani, A.K.; Wilcox, E.R.; Petit, C. OTOF Encodes Multiple Long and Short Isoforms: Genetic Evidence That the Long Ones Underlie Recessive Deafness DFNB9. Am. J. Hum. Genet. 2000, 67, 591–600. [Google Scholar] [CrossRef] [Green Version]
- Choi, B.Y.; Ahmed, Z.M.; Riazuddin, S.; Bhinder, M.A.; Shahzad, M.; Husnain, T.; Riazuddin, S.; Griffith, A.J.; Friedman, T.B. Identities and frequencies of mutations of the otoferlin gene (OTOF) causing DFNB9 deafness in Pakistan. Clin. Genet. 2009, 75, 237–243. [Google Scholar] [CrossRef] [Green Version]
- Varga, R.; Kelley, P.M.; Keats, B.J.; Starr, A.; Leal, S.M.; Cohn, E.; Kimberling, W.J. Non-syndromic recessive auditory neuropathy is the result of mutations in the otoferlin (OTOF) gene. J. Med. Genet. 2003, 40, 45–50. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Ballesteros, M.; del Castillo, F.J.; Martín, Y.; Moreno-Pelayo, M.A.; Morera, C.; Prieto, F.; Marco, J.; Morant, A.; Gallo-Terán, J.; Morales-Angulo, C.; et al. Auditory neuropathy in patients carrying mutations in the otoferlin gene (OTOF). Hum. Mutat. 2003, 22, 451–456. [Google Scholar] [CrossRef]
- Rodríguez-Ballesteros, M.; Reynoso, R.; Olarte, M.; Villamar, M.; Morera, C.; Santarelli, R.; Arslan, E.; Medá, C.; Curet, C.; Völter, C.; et al. A multicenter study on the prevalence and spectrum of mutations in the otoferlin gene (OTOF) in subjects with nonsyndromic hearing impairment and auditory neuropathy. Hum. Mutat. 2008, 29, 823–831. [Google Scholar] [CrossRef] [PubMed]
- Kitao, K.; Mutai, H.; Namba, K.; Morimoto, N.; Nakano, A.; Arimoto, Y.; Sugiuchi, T.; Masuda, S.; Okamoto, Y.; Morita, N.; et al. Deterioration in Distortion Product Otoacoustic Emissions in Auditory Neuropathy Patients With Distinct Clinical and Genetic Backgrounds. Ear Hear. 2019, 40, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Santarelli, R.; Del Castillo, I.; Cama, E.; Scimemi, P.; Starr, A. Audibility, speech perception and processing of temporal cues in ribbon synaptic disorders due to OTOF mutations. Hear. Res. 2015, 330, 200–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Lan, L.; Shi, W.; Yu, L.; Xie, L.-Y.; Xiong, F.; Zhao, C.; Li, N.; Yin, Z.; Zong, L.; et al. Temperature sensitive auditory neuropathy. Hear. Res. 2016, 335, 53–63. [Google Scholar] [CrossRef]
- Mishra, S.K.; Panda, M.R. Rapid auditory learning of temporal gap detection. J. Acoust. Soc. Am. 2016, 140, EL50. [Google Scholar] [CrossRef] [Green Version]
- Michalewski, H.J.; Starr, A.; Nguyen, T.T.; Kong, Y.Y.; Zeng, F.G. Auditory temporal processes in normal-hearing individuals and in patients with auditory neuropathy. Clin. Neurophysiol. 2005, 116, 669–680. [Google Scholar] [CrossRef] [Green Version]
- Radziwon, K.E.; June, K.M.; Stolzberg, D.J.; Xu-Friedman, M.A.; Salvi, R.J.; Dent, M.L. Behaviorally measured audiograms and gap detection thresholds in CBA/CaJ mice. J. Comp. Physiol. A 2009, 195, 961–969. [Google Scholar] [CrossRef] [Green Version]
- Santarelli, R.; Del Castillo, I.; Rodríguez-Ballesteros, M.; Scimemi, P.; Cama, E.; Arslan, E.; Starr, A. Abnormal cochlear potentials from deaf patients with mutations in the otoferlin gene. J. Assoc. Res. Otolaryngol. JARO 2009, 10, 545–556. [Google Scholar] [CrossRef] [Green Version]
- Pappa, A.K.; Hutson, K.A.; Scott, W.C.; Wilson, J.D.; Fox, K.E.; Masood, M.M.; Giardina, C.K.; Pulver, S.H.; Grana, G.D.; Askew, C.; et al. Hair cell and neural contributions to the cochlear summating potential. J. Neurophysiol. 2019, 121, 2163–2180. [Google Scholar] [CrossRef]
- Dabrowski, M.; Bukowy-Bieryllo, Z.; Zietkiewicz, E. Translational readthrough potential of natural termination codons in eucaryotes—The impact of RNA sequence. RNA Biol. 2015, 12, 950–958. [Google Scholar] [CrossRef] [Green Version]
- Duman, D.; Sirmaci, A.; Cengiz, F.B.; Ozdag, H.; Tekin, M. Screening of 38 genes identifies mutations in 62% of families with nonsyndromic deafness in Turkey. Genet. Test. Mol. Biomark. 2011, 15, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Richard, E.M.; Santos-Cortez, R.L.P.; Faridi, R.; Rehman, A.U.; Lee, K.; Shahzad, M.; Acharya, A.; Khan, A.A.; Imtiaz, A.; Chakchouk, I.; et al. Global genetic insight contributed by consanguineous Pakistani families segregating hearing loss. Hum. Mutat. 2019, 40, 53–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Migliosi, V.; Modamio-Hoybjor, S.; Moreno-Pelayo, M.A.; Rodriguez-Ballesteros, M.; Villamar, M.; Telleria, D.; Menendez, I.; Moreno, F.; Del Castillo, I. Q829X, a novel mutation in the gene encoding otoferlin (OTOF), is frequently found in Spanish patients with prelingual non-syndromic hearing loss. J. Med. Genet. 2002, 39, 502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.-C.; Tsai, C.-Y.; Lin, Y.-H.; Chen, P.-Y.; Lin, P.-H.; Cheng, Y.-F.; Wu, C.-M.; Lin, Y.-H.; Lee, C.-Y.; Erdenechuluun, J.; et al. Genetic Epidemiology and Clinical Features of Hereditary Hearing Impairment in the Taiwanese Population. Genes 2019, 10, 772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sloan-Heggen, C.M.; Bierer, A.O.; Shearer, A.E.; Kolbe, D.L.; Nishimura, C.J.; Frees, K.L.; Ephraim, S.S.; Shibata, S.B.; Booth, K.T.; Campbell, C.A.; et al. Comprehensive genetic testing in the clinical evaluation of 1119 patients with hearing loss. Hum. Genet. 2016, 135, 441–450. [Google Scholar] [CrossRef] [Green Version]
- Baux, D.; Vaché, C.; Blanchet, C.; Willems, M.; Baudoin, C.; Moclyn, M.; Faugère, V.; Touraine, R.; Isidor, B.; Dupin-Deguine, D.; et al. Combined genetic approaches yield a 48% diagnostic rate in a large cohort of French hearing-impaired patients. Sci. Rep. 2017, 7, 16783. [Google Scholar] [CrossRef] [Green Version]
- Iwasa, Y.-I.; Nishio, S.-Y.; Sugaya, A.; Kataoka, Y.; Kanda, Y.; Taniguchi, M.; Nagai, K.; Naito, Y.; Ikezono, T.; Horie, R.; et al. OTOF mutation analysis with massively parallel DNA sequencing in 2,265 Japanese sensorineural hearing loss patients. PLoS ONE 2019, 14, e0215932. [Google Scholar] [CrossRef]
- Mahdieh, N.; Shirkavand, A.; Rabbani, B.; Tekin, M.; Akbari, B.; Akbari, M.T.; Zeinali, S. Screening of OTOF mutations in Iran: A novel mutation and review. Int. J. Pediatr. Otorhinolaryngol. 2012, 76, 1610–1615. [Google Scholar] [CrossRef]
- Delmaghani, S.; del Castillo, F.J.; Michel, V.; Leibovici, M.; Aghaie, A.; Ron, U.; Van Laer, L.; Ben-Tal, N.; Van Camp, G.; Weil, D.; et al. Mutations in the gene encoding pejvakin, a newly identified protein of the afferent auditory pathway, cause DFNB59 auditory neuropathy. Nat. Genet. 2006, 38, 770–778. [Google Scholar] [CrossRef]
- Amati-Bonneau, P.; Guichet, A.; Olichon, A.; Chevrollier, A.; Viala, F.; Miot, S.; Ayuso, C.; Odent, S.; Arrouet, C.; Verny, C.; et al. OPA1 R445H mutation in optic atrophy associated with sensorineural deafness. Ann. Neurol. 2005, 58, 958–963. [Google Scholar] [CrossRef]
- Starr, A.; Isaacson, B.; Michalewski, H.J.; Zeng, F.-G.; Kong, Y.-Y.; Beale, P.; Paulson, G.W.; Keats, B.J.B.; Lesperance, M.M. A Dominantly Inherited Progressive Deafness Affecting Distal Auditory Nerve and Hair Cells. J. Assoc. Res. Otolaryngol. 2004, 5, 411–426. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.B.; Isaacson, B.; Sivakumaran, T.A.; Starr, A.; Keats, B.J.B.; Lesperance, M.M. A gene responsible for autosomal dominant auditory neuropathy (AUNA1) maps to 13q14–21. J. Med. Genet. 2004, 41, 872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, X.; Li, L.; Brashears, S.; Morlet, T.; Ng, S.S.; Berlin, C.; Hood, L.; Keats, B. Connexin 26 variants and auditory neuropathy/dys-synchrony among children in schools for the deaf. Am. J. Med Genet. Part A 2005, 139A, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Santarelli, R.; Cama, E.; Scimemi, P.; Monte, E.D.; Genovese, E.; Arslan, E. Audiological and electrocochleography findings in hearing-impaired children with connexin 26 mutations and otoacoustic emissions. Eur. Arch. Oto-Rhino-Laryngol. 2008, 265, 43–51. [Google Scholar] [CrossRef]
- Del Castillo, F.J.; Del Castillo, I. Genetics of isolated auditory neuropathies. Front. Biosci. 2012, 17, 1251–1265. [Google Scholar] [CrossRef]
- Matsunaga, T.; Mutai, H.; Kunishima, S.; Namba, K.; Morimoto, N.; Shinjo, Y.; Arimoto, Y.; Kataoka, Y.; Shintani, T.; Morita, N.; et al. A prevalent founder mutation and genotype-phenotype correlations of OTOF in Japanese patients with auditory neuropathy. Clin. Genet. 2012, 82, 425–432. [Google Scholar] [CrossRef]
- Chiu, Y.-H.; Wu, C.-C.; Lu, Y.-C.; Chen, P.-J.; Lee, W.-Y.; Liu, A.Y.-Z.; Hsu, C.-J. Mutations in the OTOF gene in Taiwanese patients with auditory neuropathy. Audiol. Neurootol. 2010, 15, 364–374. [Google Scholar] [CrossRef]
- Zhang, Q.-J.; Han, B.; Lan, L.; Zong, L.; Shi, W.; Wang, H.-Y.; Xie, L.-Y.; Wang, H.; Zhao, C.; Zhang, C.; et al. High frequency of OTOF mutations in Chinese infants with congenital auditory neuropathy spectrum disorder. Clin. Genet. 2016, 90, 238–246. [Google Scholar] [CrossRef]
- Wang, D.-Y.; Wang, Y.-C.; Weil, D.; Zhao, Y.-L.; Rao, S.-Q.; Zong, L.; Ji, Y.-B.; Liu, Q.; Li, J.-Q.; Yang, H.-M.; et al. Screening mutations of OTOF gene in Chinese patients with auditory neuropathy, including a familial case of temperature-sensitive auditory neuropathy. BMC Med. Genet. 2010, 11, 79. [Google Scholar] [CrossRef] [Green Version]
- Cho, Y.; Gong, T.-W.L.; Stöver, T.; Lomax, M.I.; Altschuler, R.A. Gene expression profiles of the rat cochlea, cochlear nucleus, and inferior colliculus. J. Assoc. Res. Otolaryngol. JARO 2002, 3, 54–67. [Google Scholar] [CrossRef] [Green Version]
- Cai, T.; Jen, H.-I.; Kang, H.; Klisch, T.J.; Zoghbi, H.Y.; Groves, A.K. Characterization of the transcriptome of nascent hair cells and identification of direct targets of the Atoh1 transcription factor. J. Neurosci. Off. J. Soc. Neurosci. 2015, 35, 5870–5883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranum, P.T.; Goodwin, A.T.; Yoshimura, H.; Kolbe, D.L.; Walls, W.D.; Koh, J.-Y.; He, D.Z.Z.; Smith, R.J.H. Insights into the Biology of Hearing and Deafness Revealed by Single-Cell RNA Sequencing. Cell Rep. 2019, 26, 3160–3171.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wynne, D.P.; Zeng, F.-G.; Bhatt, S.; Michalewski, H.J.; Dimitrijevic, A.; Starr, A. Loudness adaptation accompanying ribbon synapse and auditory nerve disorders. Brain J. Neurol. 2013, 136, 1626–1638. [Google Scholar] [CrossRef] [Green Version]
- Varga, R.; Avenarius, M.R.; Kelley, P.M.; Keats, B.J.; Berlin, C.I.; Hood, L.J.; Morlet, T.G.; Brashears, S.M.; Starr, A.; Cohn, E.S.; et al. OTOF mutations revealed by genetic analysis of hearing loss families including a potential temperature sensitive auditory neuropathy allele. J. Med. Genet. 2006, 43, 576–581. [Google Scholar] [CrossRef] [Green Version]
- Romanos, J.; Kimura, L.; Fávero, M.L.; Izarra, F.A.R.; Auricchio, M.T.B.D.M.; Batissoco, A.C.; Lezirovitz, K.; Abreu-Silva, R.S.; Mingroni-Netto, R.C. Novel OTOF mutations in Brazilian patients with auditory neuropathy. J. Hum. Genet. 2009, 54, 382–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marlin, S.; Feldmann, D.; Nguyen, Y.; Rouillon, I.; Loundon, N.; Jonard, L.; Bonnet, C.; Couderc, R.; Garabedian, E.N.; Petit, C.; et al. Temperature-sensitive auditory neuropathy associated with an otoferlin mutation: Deafening fever! Biochem. Biophys. Res. Commun. 2010, 394, 737–742. [Google Scholar] [CrossRef] [PubMed]
- Yildirim-Baylan, M.; Bademci, G.; Duman, D.; Ozturkmen-Akay, H.; Tokgoz-Yilmaz, S.; Tekin, M. Evidence for genotype-phenotype correlation for OTOF mutations. Int. J. Pediatr. Otorhinolaryngol. 2014, 78, 950–953. [Google Scholar] [CrossRef] [Green Version]
- Helfmann, S.; Neumann, P.; Tittmann, K.; Moser, T.; Ficner, R.; Reisinger, E. The crystal structure of the C₂A domain of otoferlin reveals an unconventional top loop region. J. Mol. Biol. 2011, 406, 479–490. [Google Scholar] [CrossRef]
- Meese, S.; Cepeda, A.P.; Gahlen, F.; Adams, C.M.; Ficner, R.; Ricci, A.J.; Heller, S.; Reisinger, E.; Herget, M. Activity-Dependent Phosphorylation by CaMKIIδ Alters the Ca2+Affinity of the Multi-C2-Domain Protein Otoferlin. Front. Synaptic Neurosci. 2017, 9, 13. [Google Scholar] [CrossRef] [Green Version]
- Padmanarayana, M.; Hams, N.; Speight, L.C.; Petersson, E.J.; Mehl, R.A.; Johnson, C.P. Characterization of the lipid binding properties of Otoferlin reveals specific interactions between PI(4,5)P2 and the C2C and C2F domains. Biochemistry 2014, 53, 5023–5033. [Google Scholar] [CrossRef]
- Harsini, F.M.; Bui, A.A.; Rice, A.M.; Chebrolu, S.; Fuson, K.L.; Turtoi, A.; Bradberry, M.; Chapman, E.R.; Sutton, R.B. Structural Basis for the Distinct Membrane Binding Activity of the Homologous C2A Domains of Myoferlin and Dysferlin. J. Mol. Biol. 2019, 431, 2112–2126. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. 20 years of the SMART protein domain annotation resource. Nucleic Acids Res. 2018, 46, D493–D496. [Google Scholar] [CrossRef] [PubMed]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J.E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longo-Guess, C.; Gagnon, L.H.; Bergstrom, D.E.; Johnson, K.R. A missense mutation in the conserved C2B domain of otoferlin causes deafness in a new mouse model of DFNB9. Hear. Res. 2007, 234, 21–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiménez, J.L.; Bashir, R. In silico functional and structural characterisation of ferlin proteins by mapping disease-causing mutations and evolutionary information onto three-dimensional models of their C2 domains. J. Neurol. Sci. 2007, 260, 114–123. [Google Scholar] [CrossRef]
- Harsini, F.M.; Chebrolu, S.; Fuson, K.L.; White, M.A.; Rice, A.M.; Sutton, R.B. FerA is a Membrane-Associating Four-Helix Bundle Domain in the Ferlin Family of Membrane-Fusion Proteins. Sci. Rep. 2018, 8, 10949. [Google Scholar] [CrossRef] [Green Version]
- Vilardi, F.; Stephan, M.; Clancy, A.; Janshoff, A.; Schwappach, B. WRB and CAML are necessary and sufficient to mediate tail-anchored protein targeting to the ER membrane. PLoS ONE 2014, 9, e85033. [Google Scholar] [CrossRef]
- Vogl, C.; Panou, I.; Yamanbaeva, G.; Wichmann, C.; Mangosing, S.J.; Vilardi, F.; Indzhykulian, A.A.; Pangršič, T.; Santarelli, R.; Rodriguez-Ballesteros, M.; et al. Tryptophan-rich basic protein (WRB) mediates insertion of the tail-anchored protein otoferlin and is required for hair cell exocytosis and hearing. EMBO J. 2016, 35, 2536–2552. [Google Scholar] [CrossRef] [Green Version]
- Reisinger, E. Dual-AAV delivery of large gene sequences to the inner ear. Hear. Res. 2019, 394, 107857. [Google Scholar] [CrossRef]
- Al-Moyed, H.; Cepeda, A.P.; Jung, S.; Moser, T.; Kügler, S.; Reisinger, E. A dual-AAV approach restores fast exocytosis and partially rescues auditory function in deaf otoferlin knock-out mice. EMBO Mol. Med. 2019, 11, e9396. [Google Scholar] [CrossRef]
- Akil, O.; Dyka, F.; Calvet, C.; Emptoz, A.; Lahlou, G.; Nouaille, S.; De Monvel, J.B.; Hardelin, J.-P.; Hauswirth, W.W.; Avan, P.; et al. Dual AAV-mediated gene therapy restores hearing in a DFNB9 mouse model. Proc. Natl. Acad. Sci. USA 2019, 116, 4496–4501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Starr, A.; Picton, T.W.; Sininger, Y.; Hood, L.J.; Berlin, C.I. Auditory neuropathy. Brain 1996, 119, 741–754. [Google Scholar] [CrossRef] [PubMed]
- Rouillon, I.; Marcolla, A.; Roux, I.; Marlin, S.; Feldmann, D.; Couderc, R.; Jonard, L.; Petit, C.; Denoyelle, F.; Garabédian, E.N.; et al. Results of cochlear implantation in two children with mutations in the OTOF gene. Int. J. Pediatr. Otorhinolaryngol. 2006, 70, 689–696. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vona, B.; Rad, A.; Reisinger, E. The Many Faces of DFNB9: Relating OTOF Variants to Hearing Impairment. Genes 2020, 11, 1411. https://doi.org/10.3390/genes11121411
Vona B, Rad A, Reisinger E. The Many Faces of DFNB9: Relating OTOF Variants to Hearing Impairment. Genes. 2020; 11(12):1411. https://doi.org/10.3390/genes11121411
Chicago/Turabian StyleVona, Barbara, Aboulfazl Rad, and Ellen Reisinger. 2020. "The Many Faces of DFNB9: Relating OTOF Variants to Hearing Impairment" Genes 11, no. 12: 1411. https://doi.org/10.3390/genes11121411
APA StyleVona, B., Rad, A., & Reisinger, E. (2020). The Many Faces of DFNB9: Relating OTOF Variants to Hearing Impairment. Genes, 11(12), 1411. https://doi.org/10.3390/genes11121411