Systematic Review of Sequencing Studies and Gene Expression Profiling in Familial Meniere Disease




Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Design
2.2. Research Question and Selection Criteria
- Population: Patients diagnosed with FMD.
- Intervention: Sequencing studies (Sanger or exome/genome sequencing) in FMD searching for rare variants to target candidate genes.
- Outcome: Genetic findings and pathogenicity scores reported (rare variants, candidate genes).
- Study design: Familial segregation studies.
2.3. Search Strategies
- Gene transcripts identified in the Neuropil and Somata layers of CA1 region in the Hippocampus in Rattus norvegicus by Cajigas et al. [23].
- Human synaptic genes in SynaptomeDB [24].
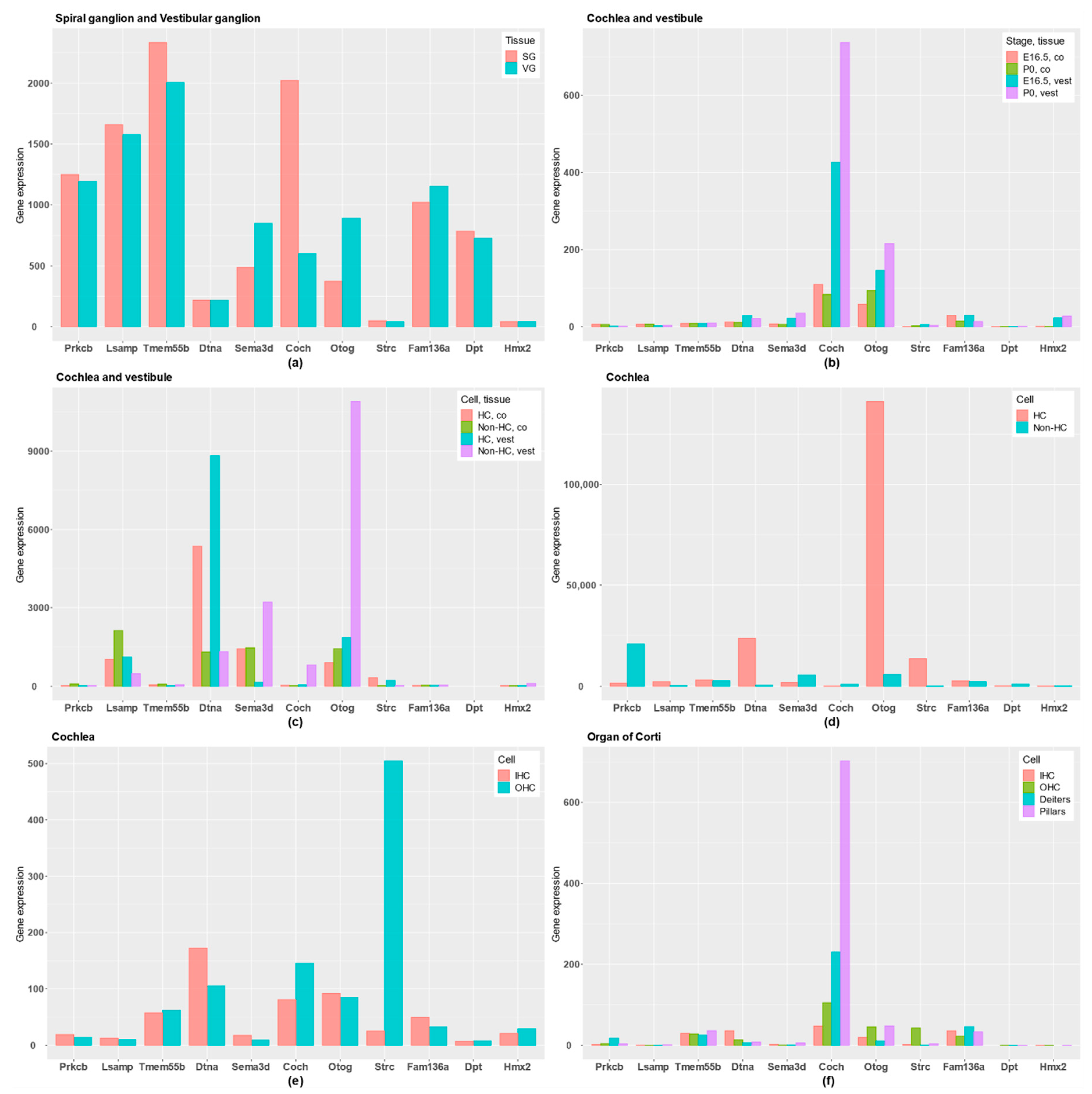
- Transcriptome catalogue of adult human inner ear, and the list of preferentially expressed mRNA genes in the inner ear when compared to 32 other tissues [25].
- RNA-Seq in embryonic day 16.5 (E16.5) and postnatal day 0 (P0) from cochlear and vestibular sensory epithelium in mouse [26].
- RNA-Seq in P0 from cochlea and vestibule in mouse, where HCs were compared with epithelial non-HCs [27].
- RNA-Seq in P0 from cochlea in mouse to contrast HC with the rest of cochlear duct [28].
- RNA-Seq in adult mice from organ of Corti, comparing inner HCs (IHC), outer HC (OHCs), Deiters’ cells and pillar cells [29].
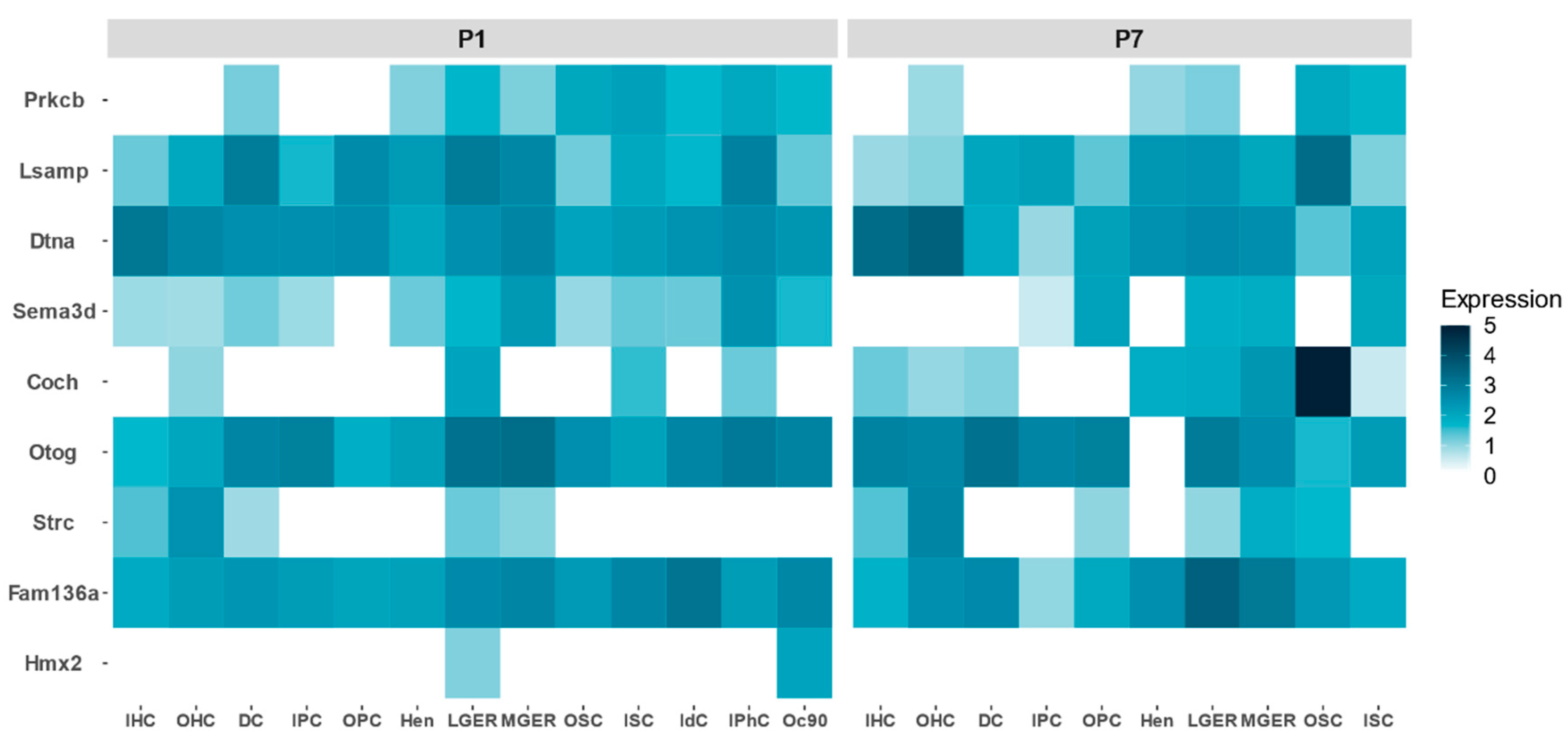
- Single cell RNA-Seq in postnatal day 1 (P1) and 7 (P7) from organ of Corti in mouse [30].
2.4. Exclusion Criteria
- Animal studies were excluded from the first analysis.
- Studies not published in English.
2.5. Quality Assessment of Selected Studies
- Is the study performed with two or more members of a family diagnosed with MD or with patients from different families but all of them diagnosed with FMD?
- Has the study reported a gene or a position in the genome statistically significant when it was compared to genome reference datasets?
- Has the study used an accurate methodology and is it described with enough details to validate its findings?
2.6. Data Extraction and Synthesis
3. Results
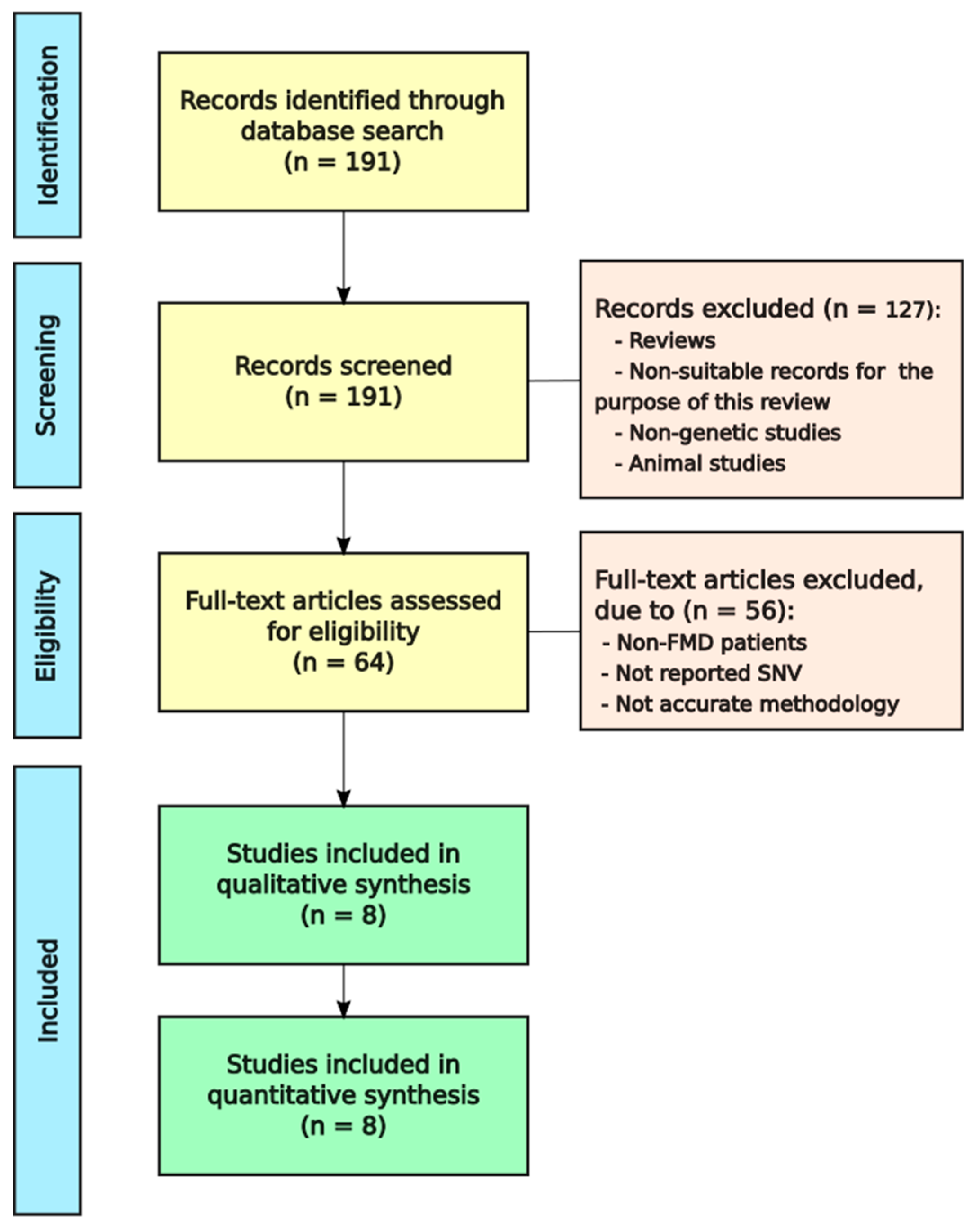
3.1. Selection and Characteristics of FMD Studies
3.2. Inheritance of SNVs Associated with FMD
3.3. Classification of Genes
4. Discussion
4.1. Main Findings in FMD Candidate Genes
4.2. Inheritance Pattern in FMD
4.3. Classification of Genes
4.4. Limitations of Systematic Review in FMD Studies
5. Conclusions
- The inheritance pattern in FMD can be AD or AR.
- Although 11 candidate genes have been reported in FMD, these genes need replication in new families and imaging studies to define which cell types are involved; they could be classified according to the gene expression profile in neural or inner ear tissues genes.
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kwan, K.Y.; Shen, J.; Corey, D.P. C-MYC Transcriptionally Amplifies SOX2 Target Genes to Regulate Self-Renewal in Multipotent Otic Progenitor Cells. Stem Cell Rep. 2015, 4, 47–60. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.C.; Appler, J.M.; Houseman, E.A.; Goodrich, L.V. Developmental profiling of spiral ganglion neurons reveals insights into auditory circuit assembly. J. Neurosci. 2011, 31, 10903–10918. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Escamez, J.A.; Carey, J.; Chung, W.-H.; Goebel, J.A.; Magnusson, M.; Mandalà, M.; Newman-Toker, D.E.; Strupp, M.; Suzuki, M.; Trabalzini, F.; et al. Diagnostic criteria for Menière’s disease. J. Vestib. Res. 2015, 25, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Escamez, J.A.; Dlugaiczyk, J.; Jacobs, J.; Lempert, T.; Teggi, R.; von Brevern, M.; Bisdorff, A. Accompanying symptoms overlap during attacks in menière’s disease and vestibular migraine. Front. Neurol. 2014, 5. [Google Scholar] [CrossRef] [Green Version]
- Hallpike, C.S.; Cairns, H. Observations on the pathology of ménière’s syndrome. J. Laryngol. Otol. 1938, 53, 625–655. [Google Scholar] [CrossRef] [Green Version]
- Alexander, T.H.; Harris, J.P. Current epidemiology of meniere’s syndrome. Otolaryngol. Clin. N. Am. 2010, 43, 965–970. [Google Scholar] [CrossRef]
- Tyrrell, J.S.; Whinney, D.J.D.; Ukoumunne, O.C.; Fleming, L.E.; Osborne, N.J. Prevalence, Associated Factors, and Comorbid Conditions for Ménière’s Disease. Ear Hear. 2014, 35, e162. [Google Scholar] [CrossRef] [PubMed]
- Frejo, L.; Martin-Sanz, E.; Teggi, R.; Trinidad, G.; Soto-Varela, A.; Santos-Perez, S.; Manrique, R.; Perez, N.; Aran, I.; Almeida-Branco, M.S.; et al. Extended phenotype and clinical subgroups in unilateral Meniere disease: A cross-sectional study with cluster analysis. Clin. Otolaryngol. 2017, 42, 1172–1180. [Google Scholar] [CrossRef] [PubMed]
- Frejo, L.; Soto-Varela, A.; Santos-Perez, S.; Aran, I.; Batuecas-Caletrio, A.; Perez-Guillen, V.; Perez-Garrigues, H.; Fraile, J.; Martin-Sanz, E.; Tapia, M.C.; et al. Clinical Subgroups in Bilateral Meniere Disease. Front. Neurol. 2016, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Requena, T.; Espinosa-Sanchez, J.M.; Cabrera, S.; Trinidad, G.; Soto-Varela, A.; Santos-Perez, S.; Teggi, R.; Perez, P.; Batuecas-Caletrio, A.; Fraile, J.; et al. Familial clustering and genetic heterogeneity in Meniere’s disease. Clin. Genet. 2014, 85, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Hietikko, E.; Kotimäki, J.; Sorri, M.; Männikkö, M. High incidence of Meniere-like symptoms in relatives of Meniere patients in the areas of Oulu University Hospital and Kainuu Central Hospital in Finland. Eur. J. Med. Genet. 2013, 56, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Klar, J.; Frykholm, C.; Friberg, U.; Dahl, N. A Meniere’s disease gene linked to chromosome 12p12.3. Am. J. Med. Genet. Part. B Neuropsychiatr. Genet. 2006, 141B, 463–467. [Google Scholar] [CrossRef] [PubMed]
- Gabriková, D.; Frykholm, C.; Friberg, U.; Lahsaee, S.; Entesarian, M.; Dahl, N.; Klar, J. Familiar Meniere’s disease restricted to 1.48 Mb on chromosome 12p12.3 by allelic and haplotype association. J. Hum. Genet. 2010, 55, 834–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hietikko, E.; Kotimäki, J.; Kentala, E.; Klockars, T.; Sorri, M.; Männikkö, M. Finnish familial Meniere disease is not linked to chromosome 12p12.3, and anticipation and cosegregation with migraine are not common findings. Genet. Med. 2011, 13, 415–420. [Google Scholar] [CrossRef] [Green Version]
- Requena, T.; Cabrera, S.; Martín-Sierra, C.; Price, S.D.; Lysakowski, A.; Lopez-Escamez, J.A. Identification of two novel mutations in FAM136A and DTNA genes in autosomal-dominant familial Meniere’s disease. Hum. Mol. Genet. 2015, 24, 1119–1126. [Google Scholar] [CrossRef]
- Martín-Sierra, C.; Requena, T.; Frejo, L.; Price, S.D.; Gallego-Martinez, A.; Batuecas-Caletrio, A.; Santos-Pérez, S.; Soto-Varela, A.; Lysakowski, A.; Lopez-Escamez, J.A. A novel missense variant in PRKCB segregates low-frequency hearing loss in an autosomal dominant family with Meniere’s disease. Hum. Mol. Genet. 2016, 25, 3407–3415. [Google Scholar] [CrossRef] [Green Version]
- Martín-Sierra, C.; Gallego-Martinez, A.; Requena, T.; Frejo, L.; Batuecas-Caletrío, A.; Lopez-Escamez, J.A. Variable expressivity and genetic heterogeneity involving DPT and SEMA3D genes in autosomal dominant familial Meniere’s disease. Eur. J. Hum. Genet. 2017, 25, 200–207. [Google Scholar] [CrossRef]
- Moher, D.; Liberati, A.; Tetzlaff, J.; Altman, D.G. Preferred reporting items for systematic reviews and meta-analyses: The PRISMA statement. J. Clin. Epidemiol. 2009, 62, 1006–1012. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Peña-Chilet, M.; Roldán, G.; Perez-Florido, J.; Ortuño, F.M.; Carmona, R.; Aquino, V.; Lopez-Lopez, D.; Loucera, C.; Fernandez-Rueda, J.L.; Gallego, A.; et al. CSVS, a crowdsourcing database of the Spanish population genetic variability. Nucleic Acids Res. 2020. [Google Scholar] [CrossRef]
- Ameur, A.; Dahlberg, J.; Olason, P.; Vezzi, F.; Karlsson, R.; Martin, M.; Viklund, J.; Kähäri, A.K.; Lundin, P.; Che, H.; et al. SweGen: A whole-genome data resource of genetic variability in a cross-section of the Swedish population. Eur. J. Hum. Genet. 2017, 25, 1253–1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karczewski, K.J.; Weisburd, B.; Thomas, B.; Solomonson, M.; Ruderfer, D.M.; Kavanagh, D.; Hamamsy, T.; Lek, M.; Samocha, K.E.; Cummings, B.B.; et al. The ExAC browser: Displaying reference data information from over 60,000 exomes. Nucleic Acids Res. 2017, 45, D840–D845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cajigas, I.J.; Tushev, G.; Will, T.J.; tom Dieck, S.; Fuerst, N.; Schuman, E.M. The local transcriptome in the synaptic neuropil revealed by deep sequencing and high-resolution imaging. Neuron 2012, 74, 453–466. [Google Scholar] [CrossRef] [Green Version]
- Pirooznia, M.; Wang, T.; Avramopoulos, D.; Valle, D.; Thomas, G.; Huganir, R.L.; Goes, F.S.; Potash, J.B.; Zandi, P.P. SynaptomeDB: An ontology-based knowledgebase for synaptic genes. Bioinformatics 2012, 28, 897–899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrauwen, I.; Hasin-Brumshtein, Y.; Corneveaux, J.J.; Ohmen, J.; White, C.; Allen, A.N.; Lusis, A.J.; Van Camp, G.; Huentelman, M.J.; Friedman, R.A. A comprehensive catalogue of the coding and non-coding transcripts of the human inner ear. Hear. Res. 2016, 333, 266–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudnicki, A.; Isakov, O.; Ushakov, K.; Shivatzki, S.; Weiss, I.; Friedman, L.M.; Shomron, N.; Avraham, K.B. Next-generation sequencing of small RNAs from inner ear sensory epithelium identifies microRNAs and defines regulatory pathways. BMC Genom. 2014, 15, 484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elkon, R.; Milon, B.; Morrison, L.; Shah, M.; Vijayakumar, S.; Racherla, M.; Leitch, C.C.; Silipino, L.; Hadi, S.; Weiss-Gayet, M.; et al. RFX transcription factors are essential for hearing in mice. Nat. Commun. 2015, 6, 8549. [Google Scholar] [CrossRef] [Green Version]
- Cai, T.; Jen, H.-I.; Kang, H.; Klisch, T.J.; Zoghbi, H.Y.; Groves, A.K. Characterization of the transcriptome of nascent hair cells and identification of direct targets of the Atoh1 transcription factor. J. Neurosci. 2015, 35, 5870–5883. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Chen, L.; Giffen, K.P.; Stringham, S.T.; Li, Y.; Judge, P.D.; Beisel, K.W.; He, D.Z.Z. Cell-specific transcriptome analysis shows that adult pillar and deiters’ cells express genes encoding machinery for specializations of cochlear hair cells. Front. Mol. Neurosci. 2018, 11, 356. [Google Scholar] [CrossRef]
- Kolla, L.; Kelly, M.C.; Mann, Z.F.; Anaya-Rocha, A.; Ellis, K.; Lemons, A.; Palermo, A.T.; So, K.S.; Mays, J.C.; Orvis, J.; et al. Characterization of the development of the mouse cochlear epithelium at the single cell level. Nat. Commun. 2020, 11. [Google Scholar] [CrossRef]
- Shen, J.; Scheffer, D.I.; Kwan, K.Y.; Corey, D.P. SHIELD: An integrative gene expression database for inner ear research. Database 2015, 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Pecka, J.L.; Zhang, Q.; Soukup, G.A.; Beisel, K.W.; He, D.Z.Z. Characterization of transcriptomes of cochlear inner and outer hair cells. J. Neurosci. 2014, 34, 11085–11095. [Google Scholar] [CrossRef] [Green Version]
- Green, R.C.; Berg, J.S.; Grody, W.W.; Kalia, S.S.; Korf, B.R.; Martin, C.L.; McGuire, A.L.; Nussbaum, R.L.; O’Daniel, J.M.; Ormond, K.E.; et al. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet. Med. 2013, 15, 565–574. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [Green Version]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef] [PubMed]
- Committee on Hearing and Equilibrium guidelines for the diagnosis and evaluation of therapy in Menière’s disease. Otolaryngol. Head Neck Surg. 1995, 113, 181–185. [CrossRef]
- Kim, B.J.; Kim, A.R.; Han, K.-H.; Rah, Y.C.; Hyun, J.; Ra, B.S.; Koo, J.-W.; Choi, B.Y. Distinct vestibular phenotypes in DFNA9 families with COCH variants. Eur. Arch. Otorhinolaryngol. 2016, 273, 2993–3002. [Google Scholar] [CrossRef] [PubMed]
- Frykholm, C.; Klar, J.; Tomanovic, T.; Ameur, A.; Dahl, N. Stereocilin gene variants associated with episodic vertigo: Expansion of the DFNB16 phenotype. Eur. J. Hum. Genet. 2018, 26, 1871–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skarp, S.; Kanervo, L.; Kotimäki, J.; Sorri, M.; Männikkö, M.; Hietikko, E. Whole-exome sequencing suggests multiallelic inheritance for childhood-onset Ménière’s disease. Ann. Hum. Genet. 2019, 83, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Roman-Naranjo, P.; Gallego-Martinez, A.; Soto-Varela, A.; Aran, I.; Moleon, M.D.C.; Espinosa-Sanchez, J.M.; Amor-Dorado, J.C.; Batuecas-Caletrio, A.; Perez-Vazquez, P.; Lopez-Escamez, J.A. Burden of Rare Variants in the OTOG Gene in Familial Meniere’s Disease. Ear Hear. 2020. [Google Scholar] [CrossRef] [PubMed]
- Mehrjoo, Z.; Kahrizi, K.; Mohseni, M.; Akbari, M.; Arzhangi, S.; Jalalvand, K.; Najmabadi, H.; Farhadi, M.; Mohseni, M.; Asghari, A.; et al. Limbic system associated membrane protein mutation in an iranian family diagnosed with ménière’s disease. Arch. Iran. Med. 2020, 23, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Gallego-Martinez, A.; Requena, T.; Roman-Naranjo, P.; May, P.; Lopez-Escamez, J.A. Enrichment of damaging missense variants in genes related with axonal guidance signalling in sporadic Meniere’s disease. J. Med. Genet. 2020, 57, 82–88. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshimura, H.; Takumi, Y.; Nishio, S.; Suzuki, N.; Iwasa, Y.; Usami, S. Deafness Gene Expression Patterns in the Mouse Cochlea Found by Microarray Analysis. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, Y.; Shirane, M.; Nakayama, K.I. TMEM55B contributes to lysosomal homeostasis and amino acid–induced mTORC1 activation. Genes Cells 2018, 23, 418–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takemasu, S.; Nigorikawa, K.; Yamada, M.; Tsurumi, G.; Kofuji, S.; Takasuga, S.; Hazeki, K. Phosphorylation of TMEM55B by Erk/MAPK regulates lysosomal positioning. J. Biochem. 2019, 166, 175–185. [Google Scholar] [CrossRef]
- Li, J.; Zhao, X.; Xin, Q.; Shan, S.; Jiang, B.; Jin, Y.; Yuan, H.; Dai, P.; Xiao, R.; Zhang, Q.; et al. Whole-exome sequencing identifies a variant in TMEM132E causing autosomal-recessive nonsyndromic hearing loss DFNB99. Hum. Mutat. 2015, 36, 98–105. [Google Scholar] [CrossRef]
- Pimenta, A.F.; Fischer, I.; Levitt, P. cDNA cloning and structural analysis of the human limbic-system-associated membrane protein (LAMP). Gene 1996, 170, 189–195. [Google Scholar] [CrossRef]
- Philips, M.-A.; Lilleväli, K.; Heinla, I.; Luuk, H.; Hundahl, C.A.; Kongi, K.; Vanaveski, T.; Tekko, T.; Innos, J.; Vasar, E. Lsamp is implicated in the regulation of emotional and social behavior by use of alternative promoters in the brain. Brain Struct. Funct. 2015, 220, 1381–1393. [Google Scholar] [CrossRef] [Green Version]
- Behan, A.T.; Byrne, C.; Dunn, M.J.; Cagney, G.; Cotter, D.R. Proteomic analysis of membrane microdomain-associated proteins in the dorsolateral prefrontal cortex in schizophrenia and bipolar disorder reveals alterations in LAMP, STXBP1 and BASP1 protein expression. Mol. Psychiatry 2009, 14, 601–613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robertson, N.G.; Cremers, C.W.R.J.; Huygen, P.L.M.; Ikezono, T.; Krastins, B.; Kremer, H.; Kuo, S.F.; Liberman, M.C.; Merchant, S.N.; Miller, C.E.; et al. Cochlin immunostaining of inner ear pathologic deposits and proteomic analysis in DFNA9 deafness and vestibular dysfunction. Hum. Mol. Genet. 2006, 15, 1071–1085. [Google Scholar] [CrossRef] [Green Version]
- Avan, P.; Gal, S.L.; Michel, V.; Dupont, T.; Hardelin, J.-P.; Petit, C.; Verpy, E. Otogelin, otogelin-like, and stereocilin form links connecting outer hair cell stereocilia to each other and the tectorial membrane. Proc. Natl. Acad. Sci. USA 2019, 116, 25948–25957. [Google Scholar] [CrossRef] [PubMed]
- Verpy, E.; Leibovici, M.; Michalski, N.; Goodyear, R.J.; Houdon, C.; Weil, D.; Richardson, G.P.; Petit, C. Stereocilin connects outer-hair-cell stereocilia to one another and to the tectorial membrane. J. Comp. Neurol 2011, 519, 194–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lien, C.F.; Mohanta, S.K.; Frontczak-Baniewicz, M.; Swinny, J.D.; Zablocka, B.; Górecki, D.C. Absence of glial α-Dystrobrevin causes abnormalities of the blood-brain barrier and progressive brain edema. J. Biol. Chem. 2012, 287, 41374–41385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aghajanian, H.; Choi, C.; Ho, V.C.; Gupta, M.; Singh, M.K.; Epstein, J.A. Semaphorin 3d and Semaphorin 3e Direct Endothelial Motility through Distinct Molecular Signaling Pathways. J. Biol. Chem. 2014, 289, 17971–17979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webber, A.; Raz, Y. Axon guidance cues in auditory development. Anat. Rec. Part A Discov. Mol. Cell. Evol. Biol. 2006, 288A, 390–396. [Google Scholar] [CrossRef]
- Requena, T.; Gallego-Martinez, A.; Lopez-Escamez, J.A. Bioinformatic integration of molecular networks and major pathways involved in mice cochlear and vestibular supporting cells. Front. Mol. Neurosci. 2018, 11. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ref. | Population | Patients | Sex | Diagnosis | Genetic Findings | |
|---|---|---|---|---|---|---|
| Gene | SNV | |||||
| [15] | Spanish | 3 | F | AAO-HNS | FAM136A | 2:70527974G>A |
| DTNA | 18:32462094G>T | |||||
| [16] | Spanish | 2 | M | AAO-HNS | PRKCB | 16:23999898G>T |
| [38] | Korean | 3 | F, M | Barany Society | COCH | 14:31349796G>A |
| [17] | Spanish | 3 | F | Barany Society | DPT | 1:168665849G>A |
| 3 | F, M | SEMA3D | 7:84642128G>A | |||
| [39] | Swedish–Norwegian | 3 | M | SNHL and episodic vertigo | STRC | 15:43896948G>A |
| [40] | Finnish | 2 | M | AAO-HNS | HMX2 | 10:124909634T>A |
| TMEM55B | 14:20927370G>A | |||||
| [41] | Spanish | 73 | F, M | Barany Society | OTOG | 11:17574758G>A |
| 11:17578774G>A | ||||||
| 11:17594747C>A | ||||||
| 11:17621218C>T | ||||||
| 11:17627548G>A | ||||||
| 11:17631453C>T | ||||||
| 11:17632921C>T | ||||||
| 11:17656672G>A | ||||||
| 11:17663747G>A | ||||||
| 11:17667139G>C | ||||||
| [42] | Iranian | 2 | F, M | Definite MD | LSAMP | 3:115561402T>C * |
| Gene | Chr | Position | ID | cDNA | Protein | Variant Effect | Allelic Frequency 1 | ACMG Classification | CADD Score | Inheritance Pattern | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| gnomAD | Other | ||||||||||
| FAM136A | 2 | 70527974 | rs690016537 | c.226C>T | p.Gln76 * | Nonsense | Novel | Pathogenic (PS3, PS4, PM2, PM4, PP3) | 41.00 | AD | |
| DTNA | 18 | 32462094 | rs533568822 | c.2143G>T | p.Val715Phe | Missense | 8.79 × 10−6 | NF (CSVS) | Pathogenic (PS3, PS4, BP1) | 24.90 | AD |
| PRKCB | 16 | 23999898 | rs1131692056 | c.275G>T | p.Gly92Val | Missense | Novel | Likely Pathogenic (PS4, PM2, PP3, PP5) | 28.20 | AD 2 | |
| COCH | 14 | 31349796 | - | - | - | - | Novel | Likely Pathogenic (PS4, PM2, PP2, PP3, PP5) | 28.10 | AD 2 | |
| DPT | 1 | 168665849 | rs748718975 | c.544C>T | p.Arg182Cys | Missense | 1.72 × 10−5 | NF (CSVS) | Likely Pathogenic (PS4, PM1, PP3, PP5, BP1) | 32.00 | AD 2 |
| SEMA3D | 7 | 84642128 | rs1057519374 | c.1738C>T | p.Pro580Ser | Missense | Novel | Pathogenic (PS4, PM1, PM2, PP3, PP5) | 25.00 | AD 2 | |
| STRC | 15 | 43896948 | rs144948296 | c.4027C>T | p.Gln1343 * | Nonsense | 3.62 × 10−4 | 0.001 (SweGen) | Pathogenic (PSV1, PS4, PM2, PP3, PP5) | 40.00 | AR |
| HMX2 | 10 | 124909634 | rs1274867386 | c.817T>A | p.Tyr273Asn | Missense | Novel | Likely Pathogenic (PS4, PM2, PP3) | 31.00 | AR 3 | |
| TMEM55B | 14 | 20927370 | rs201529818 | c.706C>T | p.Leu229Phe | Missense | 9.56 × 10−4 | 8.2 × 10−5 (ExAC) | Uncertain Significance (PS4, PP3, BS1) | 25.80 | AR 3 |
| CSVS | |||||||||||
| OTOG | 11 | 17574758 | rs552304627 | c.421G>A | p.Val141Met | Missense | 0.001288 | 0.004 | Pathogenic (PVS1, PS4, PM2, PP3, BP1) | 33.00 | AR 3 |
| 11 | 17578774 | rs61978648 | c.805G>A | p.Val269Ile | Missense | 0.004439 | 0.014 | Likely Benign (PS4, BP1, BP4, BP6) | 19.12 | AR 3 | |
| 11 | 17594747 | - | - | p.Pro747Thr | Missense | Novel | Uncertain Significance (PS4, PM2, BP1, BP4) | 21.90 | - | ||
| 11 | 17621218 | rs117005078 | c.3719C>T | p.Pro1240Leu | Missense | 0.005740 | 0.004 | Likely Pathogenic (PS4, PM2, PP3, BP1) | 33.00 | - | |
| 11 | 17627548 | rs145689709 | c.4058G>A | p.Arg1353Gln | Missense | 0.004040 | 0.006 | Uncertain Significance (PS4, PM2, BP1, BP4, BP6) | 22.00 | AR 3 | |
| 11 | 17631453 | rs117380920 | c.4642C>T | p.Leu1548Phe | Missense | 0.012350 | 0.013 | Benign (PS4, BS1, BS2, BP1, BP4, BP6) | 12.42 | - | |
| 11 | 17632921 | rs61736002 | c.6110C>T | p.Ala2037Val | Missense | 0.001207 | 0.004 | Uncertain Significance (PS4, PM2, BP1, BP4) | 7.61 | AR 3 | |
| 11 | 17656672 | rs76461792 | c.7667G>A | p.Arg2556Gln | Missense | 0.004671 | 0.004 | Benign (PS4, BS1, BS2, BP1, BP4, BP6) | 23.50 | - | |
| 11 | 17663747 | rs117315845 | c.8405G>A | p.Arg2802His | Missense | 0.002725 | 0.006 | Uncertain Significance (PS4, PM2, BP1, BP4, BP6) | 16.79 | AR 3 | |
| 11 | 17667139 | rs61997203 | c.8526G>C | p.Lys2842Asn | Missense | 0.023350 | 0.019 | Benign (PS4, BS1, BS2, BP1, BP6) | 24.20 | - | |
| LSAMP | 3 | 115561402 | - | c.673A>G | p.Lys225Glu | Missense | Novel | Likely Pathogenic (PS4, PM2) | 25.90 | AR | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Escalera-Balsera, A.; Roman-Naranjo, P.; Lopez-Escamez, J.A. Systematic Review of Sequencing Studies and Gene Expression Profiling in Familial Meniere Disease. Genes 2020, 11, 1414. https://doi.org/10.3390/genes11121414
Escalera-Balsera A, Roman-Naranjo P, Lopez-Escamez JA. Systematic Review of Sequencing Studies and Gene Expression Profiling in Familial Meniere Disease. Genes. 2020; 11(12):1414. https://doi.org/10.3390/genes11121414
Chicago/Turabian StyleEscalera-Balsera, Alba, Pablo Roman-Naranjo, and Jose Antonio Lopez-Escamez. 2020. "Systematic Review of Sequencing Studies and Gene Expression Profiling in Familial Meniere Disease" Genes 11, no. 12: 1414. https://doi.org/10.3390/genes11121414
APA StyleEscalera-Balsera, A., Roman-Naranjo, P., & Lopez-Escamez, J. A. (2020). Systematic Review of Sequencing Studies and Gene Expression Profiling in Familial Meniere Disease. Genes, 11(12), 1414. https://doi.org/10.3390/genes11121414