The Future of Livestock Management: A Review of Real-Time Portable Sequencing Applied to Livestock


Abstract
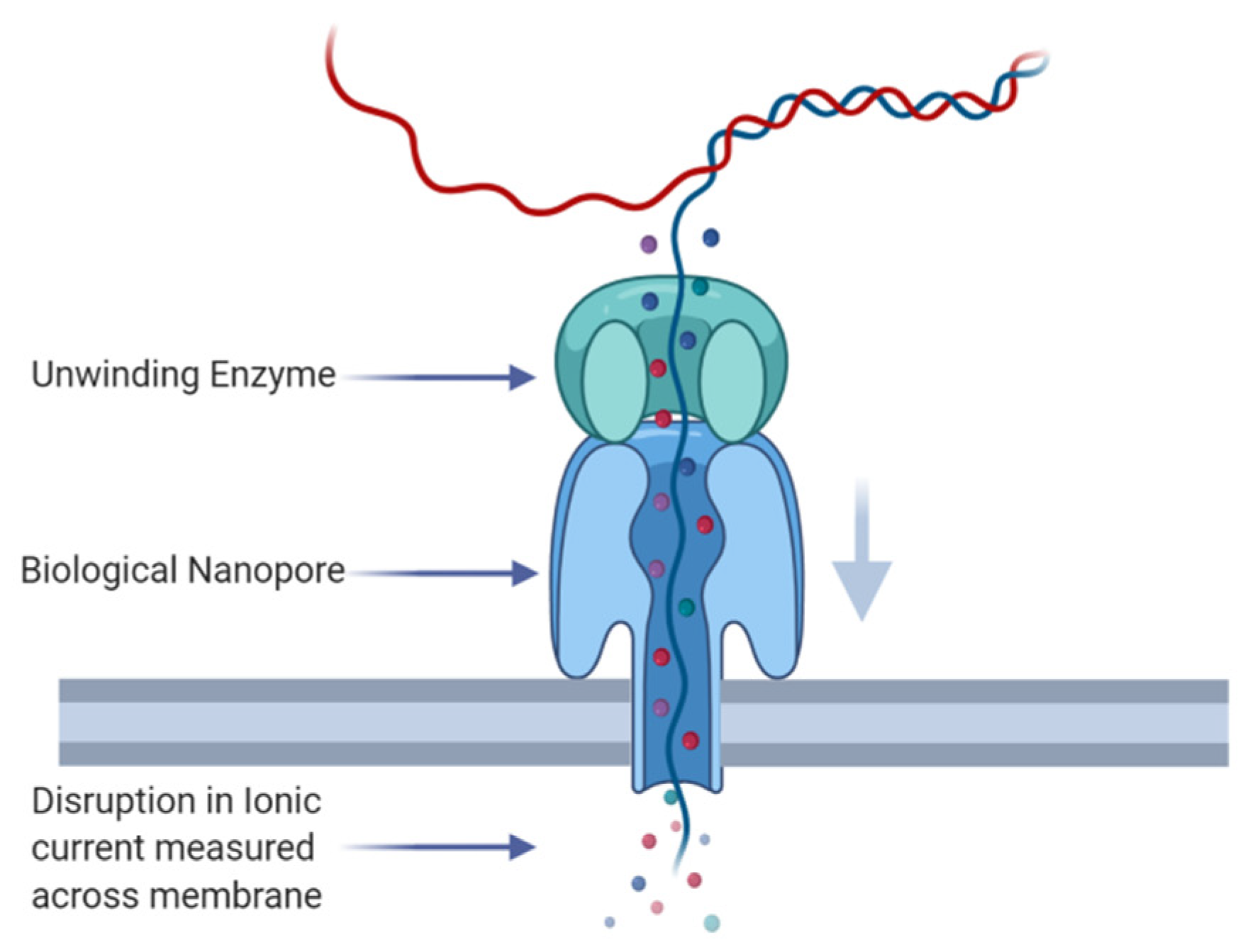
:1. Introduction
2. Current and Future Applications in Livestock
2.1. Rapid Diagnostics of Pathogens in Livestock
2.2. Reference Genomes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sequencing Platform | Read Length | Data Output | Run Time | Single-Pass Error Rate (%) | Reference |
|---|---|---|---|---|---|
| Illumina Hiseq 2000 | 2 × 150 bp | 150–200 Gb | 1–6 days | 0.16 | [70,71] |
| Illumina NextSeq 550 | 2 × 150 bp | 120 Gb | 15–18 h | 0.16 | [70,71] |
| Pacbio RS II | 10–15 kb on average | 0.5–1 Gb | 0.5–4 h | 10 | [16,71] |
| PacBio Sequel | 10–15 kb on average | 15 Gb | Up to 20 h | 10 | [72] |
| PacBio Sequel II | 10–15 kb on average | 80–100 Gb | Up to 30 h | 10 | [73] |
| Oxford Nanopore MinION | 10–20 kb on average | 20–30 Gb (per flow cell) Up to 1 flow cell/run | Up to 96 h | 5–20 | [12,74] |
| Oxford Nanopore GridION | 10–20 kb on average | 20–30 Gb (per flow cell) Up to 5 flow cells/run | Up to 96 h | 5–20 | [12,74] |
| Oxford Nanopore PromethION | 10–20 kb on average | 100–180 Gb (per flow cell) Up to 48 flow cells/run | Up to 96 h | 5–20 | [12,74] |

2.3. Structural Variants
2.4. DNA/RNA Modification
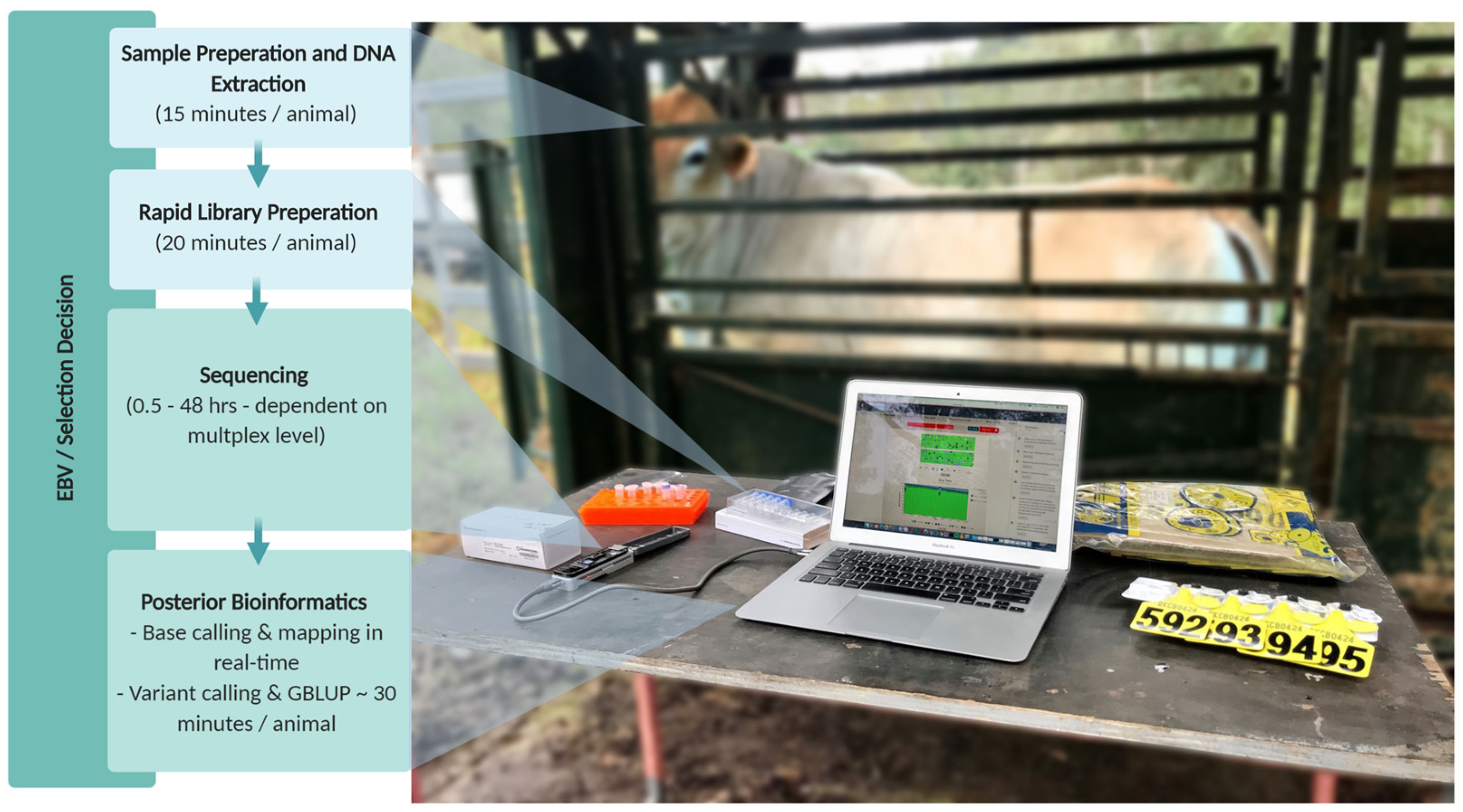
2.5. Genomic Prediction and Crush-Side Genotyping
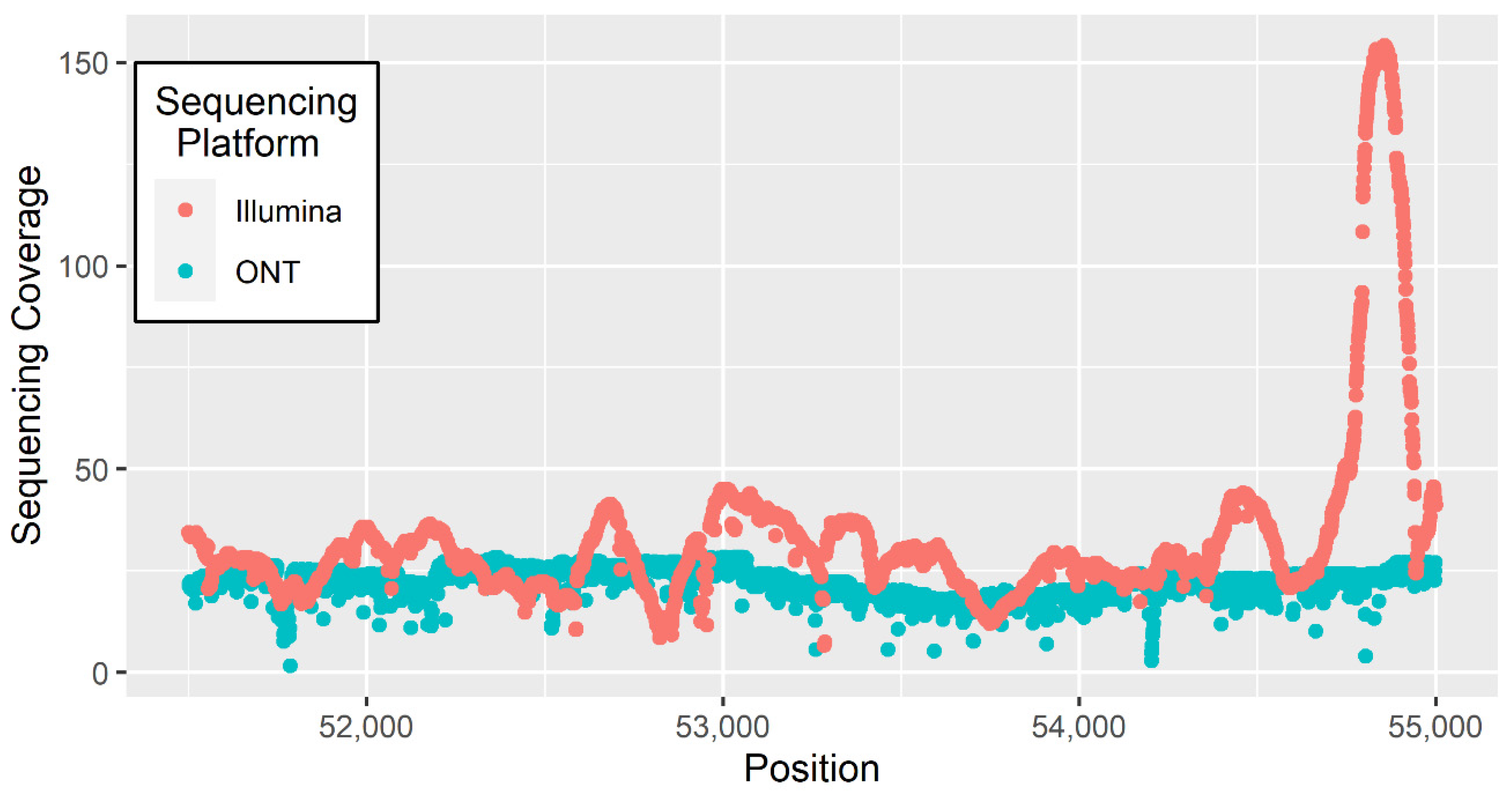
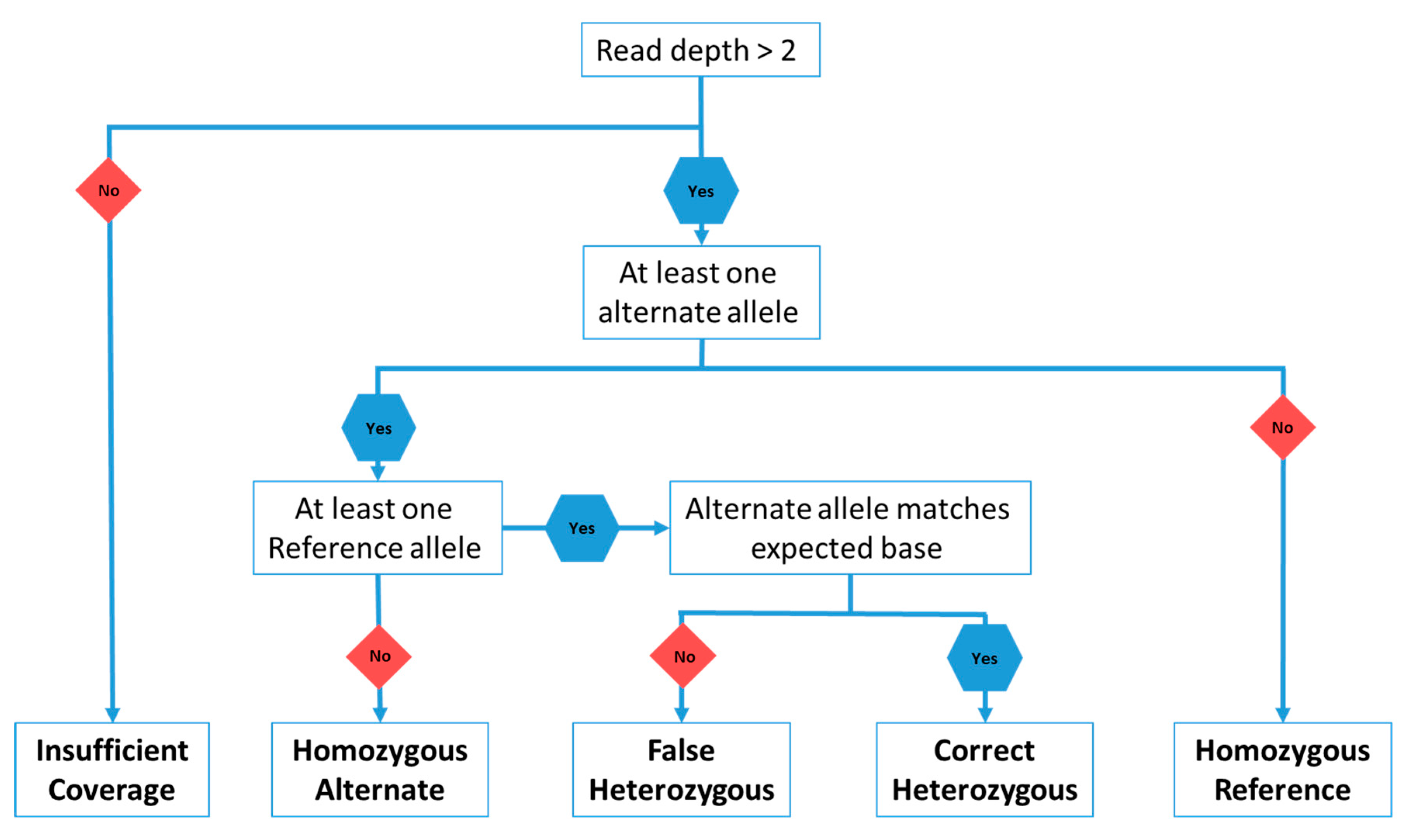
3. Materials and Methods
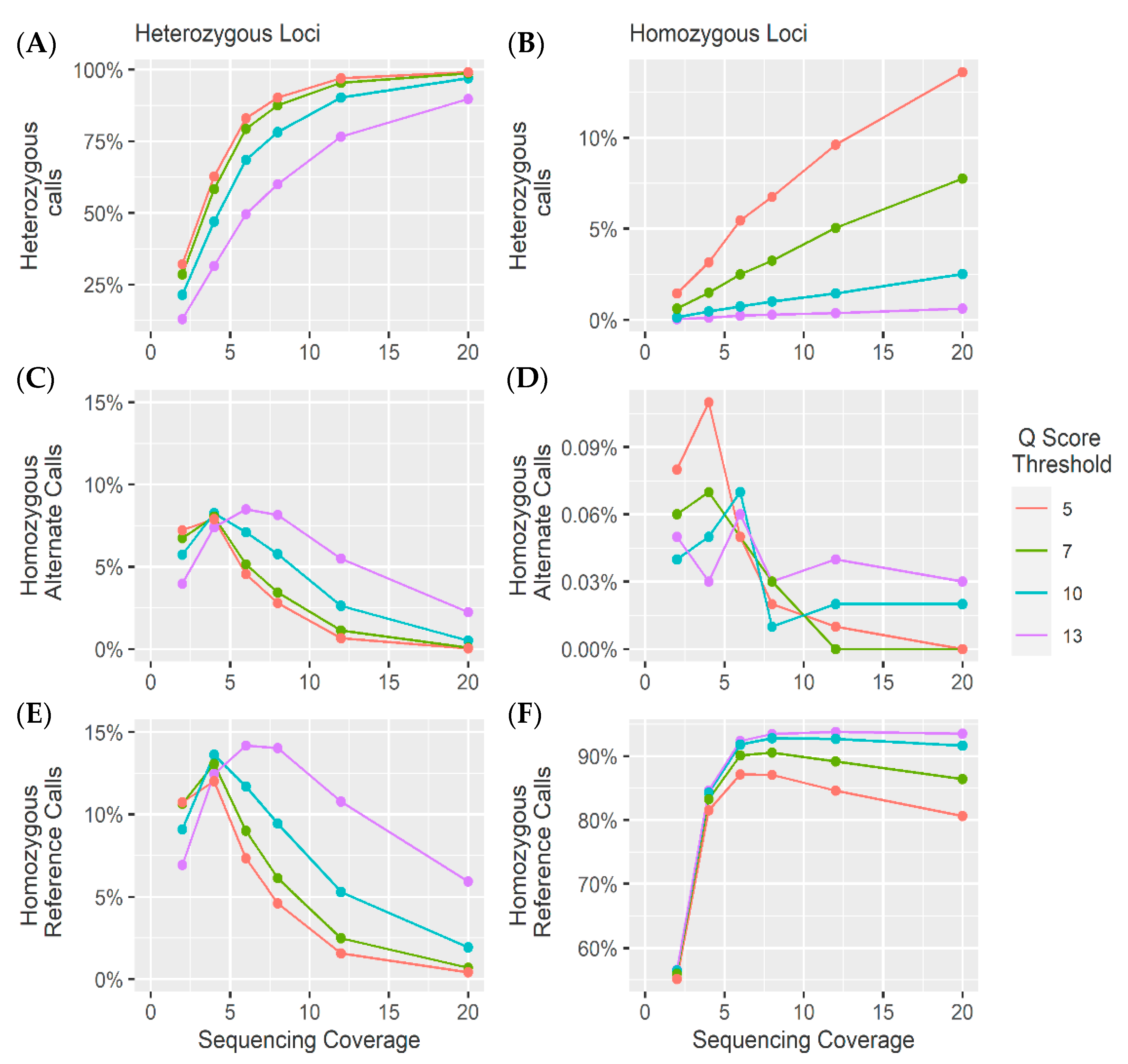
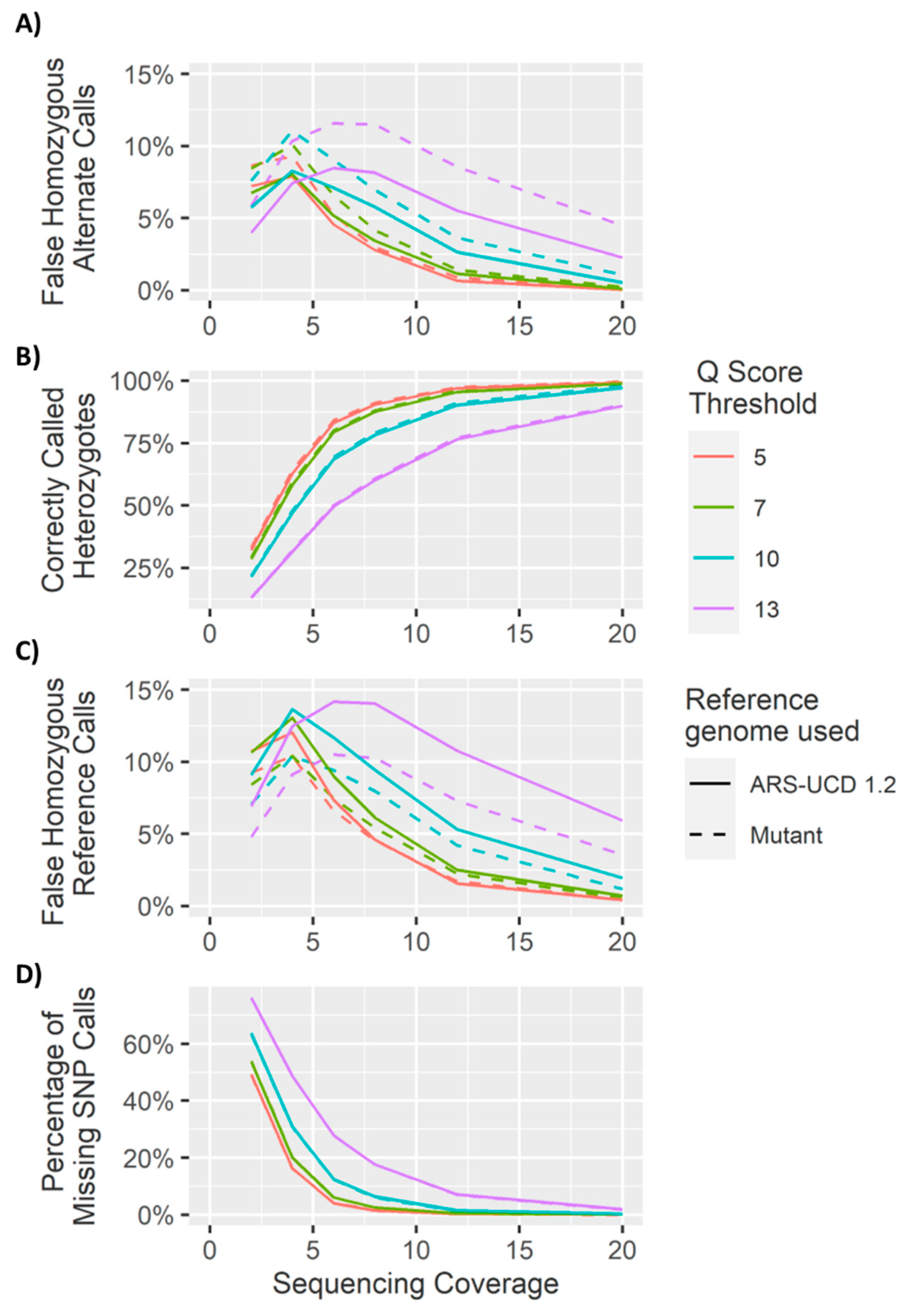
4. Results and Discussion
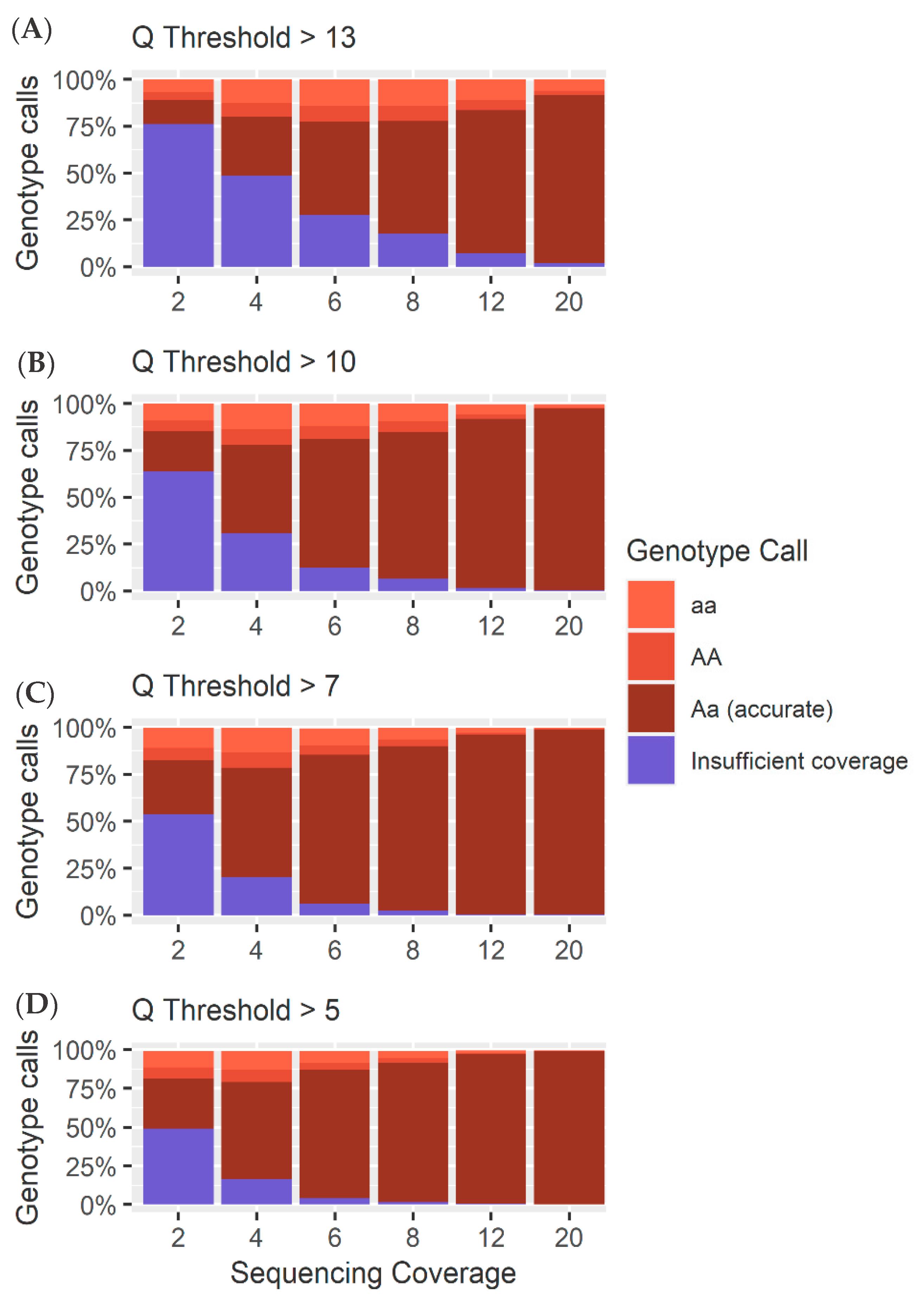
4.1. Heterozygous Positions
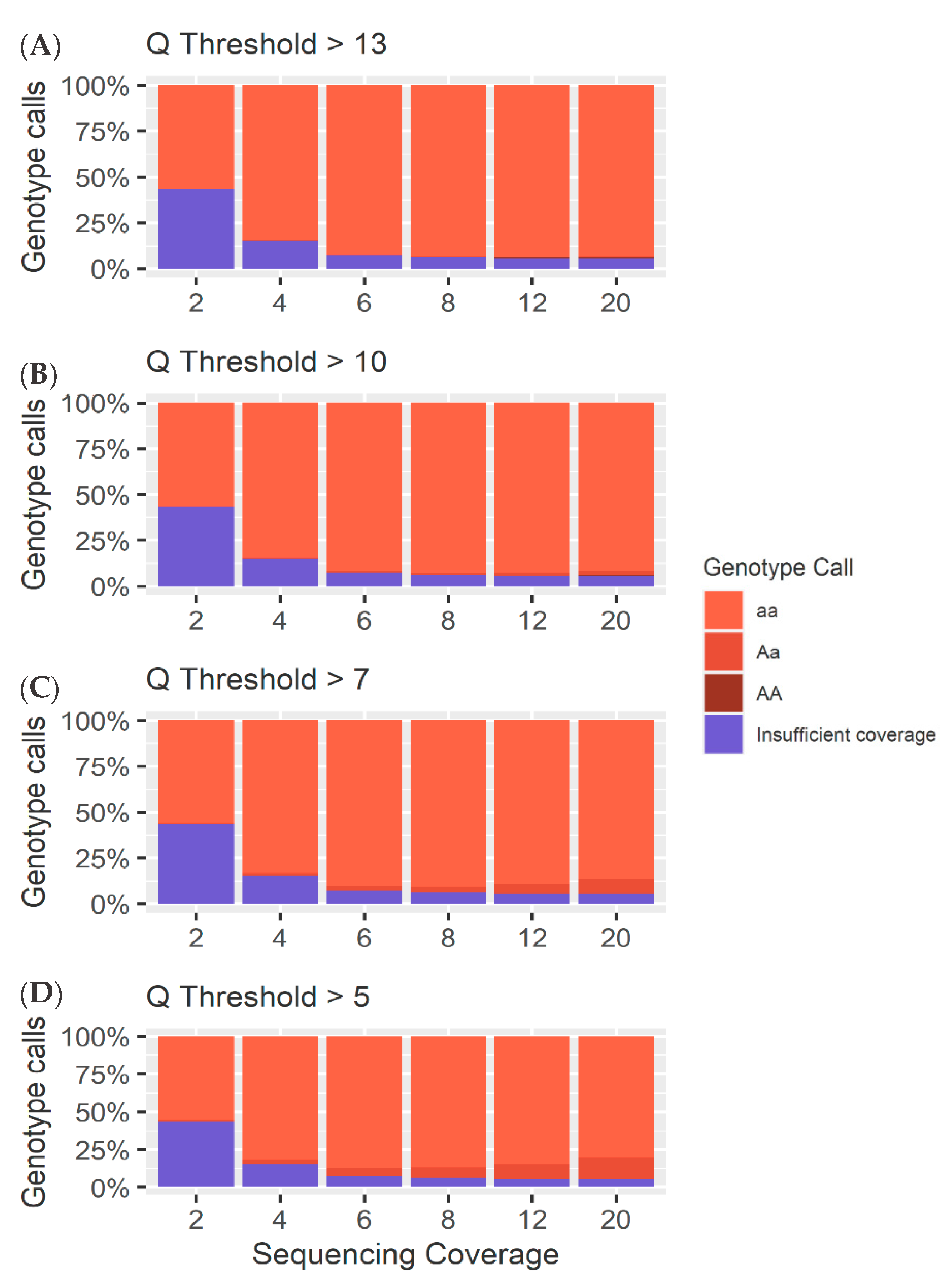
4.2. Homozygous Positions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pareek, C.S.; Smoczynski, R.; Tretyn, A. Sequencing technologies and genome sequencing. J. Appl. Genet. 2011, 52, 413–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heather, J.M.; Chain, B. The sequence of sequencers: The history of sequencing DNA. Genom. 2016, 107, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Acinas, S.G.; Sarma-Rupavtarm, R.; Klepac-Ceraj, V.; Polz, M.F. PCR-induced sequence artifacts and bias: Insights from comparison of two 16S rRNA clone libraries constructed from the same sample. J. Appl. Environ. Microbiol. 2005, 71, 8966–8969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kebschull, J.M.; Zador, A.M. Sources of PCR-induced distortions in high-throughput sequencing data sets. Nucleic Acids Res. 2015, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jansen, H.J.; Liem, M.; Jong-Raadsen, S.A.; Dufour, S.; Weltzien, F.A.; Swinkels, W.; Koelewijn, A.; Palstra, A.P.; Pelster, B.; Spaink, H.P.; et al. Author Correction: Rapid de novo assembly of the European eel genome from nanopore sequencing reads. Sci. Rep. 2019, 9, 7911. [Google Scholar] [CrossRef] [PubMed]
- Michael, T.P.; Jupe, F.; Bemm, F.; Motley, S.T.; Sandoval, J.P.; Lanz, C.; Loudet, O.; Weigel, D.; Ecker, J.R. High contiguity Arabidopsis thaliana genome assembly with a single nanopore flow cell. Nat. Commun. 2018, 9, 541. [Google Scholar] [CrossRef] [Green Version]
- Diaz-Viraque, F.; Pita, S.; Greif, G.; De Souza, R.D.M.; Iraola, G.; Robello, C. Nanopore Sequencing Significantly Improves Genome Assembly of the Protozoan Parasite Trypanosoma cruzi. Genome Biol. Evol. 2019, 11, 1952–1957. [Google Scholar] [CrossRef]
- Hunt, S.P.; Jarvis, D.E.; Larsen, D.J.; Mosyakin, S.L.; Kolano, B.A.; Jackson, E.W.; Martin, S.L.; Jellen, E.N.; Maughan, P.J. A Chromosome-Scale Assembly of the Garden Orach (Atriplex hortensis L.) Genome Using Oxford Nanopore Sequencing. Front. Plant. Sci. 2020, 11. [Google Scholar] [CrossRef]
- Liu, L.; Li, Y.; Li, S.; Hu, N.; He, Y.; Pong, R.; Lin, D.; Lu, L.; Law, M. Comparison of next-generation sequencing systems. J. Biomed. Biotechnol. 2012, 2012, 251364. [Google Scholar] [CrossRef]
- Leggett, R.M.; Clark, M.D. A world of opportunities with nanopore sequencing. J. Exp. Bot. 2017, 68, 5419–5429. [Google Scholar] [CrossRef]
- Weirather, J.L.; de Cesare, M.; Wang, Y.; Piazza, P.; Sebastiano, V.; Wang, X.J.; Buck, D.; Au, K.F. Comprehensive comparison of Pacific Biosciences and Oxford Nanopore Technologies and their applications to transcriptome analysis. F1000Research 2017, 6, 100. [Google Scholar] [CrossRef] [PubMed]
- Oxford Nanopore Technologies. Products. Available online: https://nanoporetech.com/products (accessed on 19 June 2020).
- Ashton, P.M.; Nair, S.; Dallman, T.; Rubino, S.; Rabsch, W.; Mwaigwisya, S.; Wain, J.; O’Grady, J. MinION nanopore sequencing identifies the position and structure of a bacterial antibiotic resistance island. Nat. Biotechnol. 2015, 33, 296–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Santis, D.; Truong, L.; Martinez, P.; D’Orsogna, L. Rapid high-resolution HLA genotyping by MinION Oxford nanopore sequencing for deceased donor organ allocation. HLA 2020, 96, 141–162. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.V.; Blades, N.; Ding, J.; Sultana, R.; Parmigiani, G. Estimation of sequencing error rates in short reads. BMC Bioinform. 2012, 13, 185. [Google Scholar] [CrossRef] [Green Version]
- Minoche, A.E.; Dohm, J.C.; Himmelbauer, H. Evaluation of genomic high-throughput sequencing data generated on Illumina HiSeq and genome analyzer systems. Genome Biol. 2011, 12, R112. [Google Scholar] [CrossRef] [Green Version]
- Rang, F.J.; Kloosterman, W.P.; de Ridder, J. From squiggle to basepair: Computational approaches for improving nanopore sequencing read accuracy. Genome Biol. 2018, 19, 90. [Google Scholar] [CrossRef] [Green Version]
- Weischenfeldt, J.; Symmons, O.; Spitz, F.; Korbel, J.O. Phenotypic impact of genomic structural variation: Insights from and for human disease. Nat. Rev. Genet. 2013, 14, 125–138. [Google Scholar] [CrossRef]
- Shafin, K.; Pesout, T.; Lorig-Roach, R.; Haukness, M.; Olsen, H.E.; Bosworth, C.; Armstrong, J.; Tigyi, K.; Maurer, N.; Koren, S.; et al. Nanopore sequencing and the Shasta toolkit enable efficient de novo assembly of eleven human genomes. Nat. Biotechnol. 2020, 38, 1044–1053. [Google Scholar] [CrossRef]
- James, P.; Stoddart, D.; Harrington, E.D.; Beaulaurier, J.; Ly, L.; Reid, S.; Turner, D.J.; Juul, S. LamPORE: Rapid, accurate and highly scalable molecular screening for SARS-CoV-2 infection, based on nanopore sequencing. medRxiv 2020. [Google Scholar] [CrossRef]
- Samarakoon, H.; Punchihewa, S.; Senanayake, A.; Ragel, R.; Gamaarachchi, H. F5N: Nanopore Sequence Analysis Toolkit for Android Smartphones. bioRxiv 2020. [Google Scholar] [CrossRef]
- Mitsuhashi, S.; Kryukov, K.; Nakagawa, S.; Takeuchi, J.S.; Shiraishi, Y.; Asano, K.; Imanishi, T. A portable system for rapid bacterial composition analysis using a nanopore-based sequencer and laptop computer. Sci. Rep.-UK 2017, 7, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Quick, J.; Ashton, P.; Calus, S.; Chatt, C.; Gossain, S.; Hawker, J.; Nair, S.; Neal, K.; Nye, K.; Peters, T.; et al. Rapid draft sequencing and real-time nanopore sequencing in a hospital outbreak of Salmonella. Genome Biol. 2015, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greninger, A.L.; Naccache, S.N.; Federman, S.; Yu, G.X.; Mbala, P.; Bres, V.; Stryke, D.; Bouquet, J.; Somasekar, S.; Linnen, J.M.; et al. Rapid metagenomic identification of viral pathogens in clinical samples by real-time nanopore sequencing analysis. Genome Med. 2015, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greig, D.R.; Jenkins, C.; Gharbia, S.; Dallman, T.J. Comparison of single-nucleotide variants identified by Illumina and Oxford Nanopore technologies in the context of a potential outbreak of Shiga toxin-producing Escherichia coli. Gigascience 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Bialasiewicz, S.; Duarte, T.P.S.; Nguyen, S.H.; Sukumaran, V.; Stewart, A.; Appleton, S.; Pitt, M.E.; Bainomugisa, A.; Jennison, A.V.; Graham, R.; et al. Rapid diagnosis of Capnocytophaga canimorsus septic shock in an immunocompetent individual using real-time Nanopore sequencing: A case report. BMC Infect. Dis. 2019, 19. [Google Scholar] [CrossRef] [Green Version]
- Quick, J.; Loman, N.J.; Duraffour, S.; Simpson, J.T.; Ettore, S.; Cowley, L.; Bore, J.A.; Koundouno, R.; Dudas, G.; Mikhail, A.; et al. Real-time, portable genome sequencing for Ebola surveillance. Nature 2016, 530, 228. [Google Scholar] [CrossRef]
- Giovanetti, M.; Faria, N.R.; Lourenco, J.; de Jesus, J.G.; Xavier, J.; Claro, I.M.; Kraemer, M.U.G.; Fonseca, V.; Dellicour, S.; Theze, J.; et al. Genomic and Epidemiological Surveillance of Zika Virus in the Amazon Region. Cell Rep. 2020, 30, 2275–2283. [Google Scholar] [CrossRef] [Green Version]
- Taiaroa, G.; Rawlinson, D.; Featherstone, L.; Pitt, M.; Caly, L.; Druce, J.; Purcell, D.; Harty, L.; Tran, T.; Roberts, J.; et al. Direct RNA sequencing and early evolution of SARS-CoV-2. bioRxiv 2020. [Google Scholar] [CrossRef]
- Kim, D.; Lee, J.Y.; Yang, J.S.; Kim, J.W.; Kim, V.N.; Chang, H. The Architecture of SARS-CoV-2 Transcriptome. Cell 2020, 181, 914–921 e910. [Google Scholar] [CrossRef]
- Xu, Y.F.; Lewandowski, K.; Jeffery, K.; Downs, L.O.; Foster, D.; Sanderson, N.D.; Kavanagh, J.; Vaughan, A.; Salvagno, C.; Vipond, R.; et al. Nanopore metagenomic sequencing to investigate nosocomial transmission of human metapneumovirus from a unique genetic group among haematology patients in the United Kingdom. J. Infect. 2020, 80, 571–577. [Google Scholar] [CrossRef] [Green Version]
- Rhodes, J.; Abdolrasouli, A.; Farrer, R.A.; Cuomo, C.A.; Aanensen, D.M.; Armstrong-James, D.; Fisher, M.C.; Schelenz, S. Genomic epidemiology of the UK outbreak of the emerging human fungal pathogen Candida auris (vol 7, pg 43, 2018). Emerg. Microbes Infect. 2018, 7. [Google Scholar] [CrossRef]
- Costard, S.; Wieland, B.; de Glanville, W.; Jori, F.; Rowlands, R.; Vosloo, W.; Roger, F.; Pfeiffer, D.U.; Dixon, L.K. African swine fever: How can global spread be prevented? Philos. Trans. R. Soc. B 2009, 364, 2683–2696. [Google Scholar] [CrossRef] [Green Version]
- Galindo, I.; Alonso, C. African Swine Fever Virus: A Review. Viruses 2017, 9, 103. [Google Scholar] [CrossRef] [Green Version]
- Mason-D’Croz, D.; Bogard, J.R.; Herrero, M.; Robinson, S.; Sulser, T.B.; Wiebe, K.; Willenbockel, D.; Godfray, H.C.J. Modelling the global economic consequences of a major African swine fever outbreak in China. Nat. Food 2020, 1, 221–228. [Google Scholar] [CrossRef] [Green Version]
- Dixon, L.K.; Islam, M.; Nash, R.; Reis, A.L. African swine fever virus evasion of host defences. Virus Res. 2019, 266, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.H.; Atim, S.; LeBlanc, N.; Rauh, R.; Esau, M.; Chenais, E.; Mwebe, R.; Nelson, W.M.; Masembe, C.; Nantima, N.; et al. Overcoming the challenges of pen-side molecular diagnosis of African swine fever to support outbreak investigations under field conditions. Transbound Emerg. Dis. 2019, 66, 908–914. [Google Scholar] [CrossRef]
- O’Donnell, V.K.; Grau, F.R.; Mayr, G.A.; Samayoa, T.L.S.; Dodd, K.A.; Barrette, R.W. Rapid Sequence-Based Characterization of African Swine Fever Virus by Use of the Oxford Nanopore MinION Sequence Sensing Device and a Companion Analysis Software Tool. J. Clin. Microbiol. 2020, 58. [Google Scholar] [CrossRef] [Green Version]
- Ryan, W.; Jeremy, C.; Nick, L. PoreChop. Available online: https://github.com/rrwick/Porechop (accessed on 8 October 2020).
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Gallardo, C.; Fernandez-Pinero, J.; Pelayo, V.; Gazaev, I.; Markowska-Daniel, I.; Pridotkas, G.; Nieto, R.; Fernandez-Pacheco, P.; Bokhan, S.; Nevolko, O.; et al. Genetic Variation among African Swine Fever Genotype II Viruses, Eastern and Central Europe. Emerg. Infect. Dis. 2014, 20, 1544–1547. [Google Scholar] [CrossRef] [Green Version]
- Theuns, S.; Vanmechelen, B.; Bernaert, Q.; Deboutte, W.; Vandenhole, M.; Beller, L.; Matthijnssens, J.; Maes, P.; Nauwynck, H.J. Nanopore sequencing as a revolutionary diagnostic tool for porcine viral enteric disease complexes identifies porcine kobuvirus as an important enteric virus. Sci. Rep.-UK 2018, 8, 9830. [Google Scholar] [CrossRef] [Green Version]
- Omaleki, L.; Blackall, P.J.; Cuddihy, T.; Beatson, S.A.; Forde, B.M.; Turni, C. Using genomics to understand inter- and intra- outbreak diversity of Pasteurella multocida isolates associated with fowl cholera in meat chickens. Microb. Genom. 2020, 6, e000346. [Google Scholar] [CrossRef] [PubMed]
- Baksi, S.; Rao, N.; Khan, M.; Chauhan, P.; Chauhan, A. Efficacy of Inactivated Fowl Cholera Vaccine in Chickens. Psm. Vet. Res. 2018, 3, 32–35. [Google Scholar]
- Snowder, G.D.; Van Vleck, L.D.; Cundiff, L.V.; Bennett, G.L. Bovine respiratory disease in feedlot cattle: Environmental, genetic, and economic factors. J. Anim. Sci. 2006, 84, 1999–2008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noyes, N.R.; Benedict, K.M.; Gow, S.P.; Booker, C.W.; Hannon, S.J.; McAllister, T.A.; Morley, P.S. Mannheimia haemolytica in Feedlot Cattle: Prevalence of Recovery and Associations with Antimicrobial Use, Resistance, and Health Outcomes. J. Vet. Intern. Med. 2015, 29, 705–713. [Google Scholar] [CrossRef]
- Griffin, D.; Chengappa, M.M.; Kuszak, J.; McVey, D.S. Bacterial Pathogens of the Bovine Respiratory Disease Complex. Vet. Clin. N. Am.-Food A 2010, 26, 381–394. [Google Scholar] [CrossRef]
- Taylor, J.D.; Fulton, R.W.; Lehenbauer, T.W.; Step, D.L.; Confer, A.W. The epidemiology of bovine respiratory disease: What is the evidence for predisposing factors? Can. Vet. J. 2010, 51, 1095–1102. [Google Scholar]
- Edwards, T.A. Control methods for bovine respiratory disease for feedlot cattle. Vet. Clin. N. Am. Food Anim Pract. 2010, 26, 273–284. [Google Scholar] [CrossRef]
- McCabe, M.S.; Cormican, P.; Johnston, D.; Earley, B. Simultaneous detection of DNA and RNA virus species involved in bovine respiratory disease by PCR-free rapid tagmentation-based library preparation and MinION nanopore sequencing. bioRxiv 2018. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Huang, Y.; Godson, D.L.; Fernando, C.; Alexander, T.W.; Hill, J.E. Assessment of metagenomic sequencing and qPCR for detection of influenza D virus in bovine respiratory tract samples. bioRxiv 2020. [Google Scholar] [CrossRef]
- Shaffer, L. Inner Workings: Portable DNA sequencer helps farmers stymie devastating viruses. Proc. Natl. Acad. Sci. USA 2019, 116, 3351–3353. [Google Scholar] [CrossRef] [Green Version]
- Lim, A.; Naidenova, B.; Bates, H.; Willyerd, K.; Snider, T.; Couger, M.B.; Chen, C.; Ramachandran, A. Nanopore ultra-long read sequencing technology for antimicrobial resistance detection in Mannheimia haemolytica. J. Microbiol. Meth. 2019, 159, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Andrew, R.; James, H.; Nick, L.; Aine, O.t.; Chris, S.; Roger, P.; Botond, S. RAMPART. Available online: https://github.com/artic-network/rampart (accessed on 8 October 2020).
- Nick, L.; Will, R.; Andrew, R. nCoV-2019 Novel Coronavirus Bioinformatics Protocol. Available online: https://artic.network/ncov-2019/ncov2019-bioinformatics-sop.html (accessed on 8 October 2020).
- Payne, A.; Holmes, N.; Clarke, T.; Munro, R.; Debebe, B.; Loose, M. Nanopore adaptive sequencing for mixed samples, whole exome capture and targeted panels. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Kovaka, S.; Fan, Y.; Ni, B.; Timp, W.; Schatz, M.C. Targeted nanopore sequencing by real-time mapping of raw electrical signal with UNCALLED. bioRxiv 2020. [Google Scholar] [CrossRef]
- Houe, H. Epidemiological features and economical importance of bovine virus diarrhoea virus (BVDV) infections. Vet. Microbiol. 1999, 64, 89–107. [Google Scholar] [CrossRef]
- Alexandersen, S.; Zhang, Z.; Donaldson, A.I.; Garland, A.J. The pathogenesis and diagnosis of foot-and-mouth disease. J. Comp. Pathol. 2003, 129, 1–36. [Google Scholar] [CrossRef]
- Thornton, P.K. Livestock production: Recent trends, future prospects. Philos. Trans. R. Soc. Lond B Biol. Sci. 2010, 365, 2853–2867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bickhart, D.M.; Liu, G.E. The challenges and importance of structural variation detection in livestock. Front. Genet. 2014, 5, 37. [Google Scholar] [CrossRef] [Green Version]
- Zimin, A.V.; Delcher, A.L.; Florea, L.; Kelley, D.R.; Schatz, M.C.; Puiu, D.; Hanrahan, F.; Pertea, G.; Van Tassell, C.P.; Sonstegard, T.S.; et al. A whole-genome assembly of the domestic cow, Bos taurus. Genome Biol. 2009, 10, R42. [Google Scholar] [CrossRef] [Green Version]
- International Chicken Genome Sequencing, C. Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution. Nature 2004, 432, 695–716. [Google Scholar] [CrossRef]
- International Sheep Genomics, C.; Archibald, A.L.; Cockett, N.E.; Dalrymple, B.P.; Faraut, T.; Kijas, J.W.; Maddox, J.F.; McEwan, J.C.; Hutton Oddy, V.; Raadsma, H.W.; et al. The sheep genome reference sequence: A work in progress. Anim. Genet. 2010, 41, 449–453. [Google Scholar] [CrossRef]
- Fang, X.; Mou, Y.; Huang, Z.; Li, Y.; Han, L.; Zhang, Y.; Feng, Y.; Chen, Y.; Jiang, X.; Zhao, W.; et al. The sequence and analysis of a Chinese pig genome. Gigascience 2012, 1, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimin, A.V.; Kelley, D.R.; Roberts, M.; Marçais, G.; Salzberg, S.L.; Yorke, J.A. Mis-assembled “segmental duplications” in two versions of the Bos taurus genome. PLoS ONE 2012, 7, e42680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, D.; Zhang, S.; Ren, P.; Liang, F.; Sun, Z.; Meng, G.; Tan, Y.; Hu, J.; Li, X.; Lai, Q.; et al. Comparison of the two up-to-date sequencing technologies for genome assembly: HiFi reads of Pacbio Sequel II system and ultralong reads of Oxford Nanopore. bioRxiv 2020. [Google Scholar] [CrossRef]
- Upadhyay, M.; Hauser, A.; Kunz, E.; Krebs, S.; Blum, H.; Dotsev, A.; Okhlopkov, I.; Bagirov, V.; Brem, G.; Zinovieva, N.; et al. The first draft genome assembly of Snow sheep (Ovis nivicola). Genome Biol. Evol. 2020. [Google Scholar] [CrossRef]
- Scott, A.D.; Zimin, A.V.; Puiu, D.; Workman, R.; Britton, M.; Zaman, S.; Caballero, M.; Read, A.C.; Bogdanove, A.J.; Burns, E.; et al. The giant sequoia genome and proliferation of disease resistance genes. bioRxiv 2020. [Google Scholar] [CrossRef]
- Pillai, S.; Gopalan, V.; Lam, A.K.-Y. Review of sequencing platforms and their applications in phaeochromocytoma and paragangliomas. Crit. Rev. Oncol. Hematol. 2017, 116, 58–67. [Google Scholar] [CrossRef]
- Rhoads, A.; Au, K.F. PacBio Sequencing and Its Applications. Genom. Proteom. Bioinformt. 2015, 13, 278–289. [Google Scholar] [CrossRef] [Green Version]
- Loit, K.; Adamson, K.; Bahram, M.; Puusepp, R.; Anslan, S.; Kiiker, R.; Drenkhan, R.; Tedersoo, L. Relative Performance of MinION (Oxford Nanopore Technologies) versus Sequel (Pacific Biosciences) Third-Generation Sequencing Instruments in Identification of Agricultural and Forest Fungal Pathogens. Appl. Environ. Microbiol. 2019, 85, e01368-19. [Google Scholar] [CrossRef]
- Hon, T.; Mars, K.; Young, G.; Tsai, Y.-C.; Karalius, J.W.; Landolin, J.M.; Maurer, N.; Kudrna, D.; Hardigan, M.A.; Steiner, C.C.; et al. Highly accurate long-read HiFi sequencing data for five complex genomes. bioRxiv 2020. [Google Scholar] [CrossRef]
- Kono, N.; Arakawa, K. Nanopore sequencing: Review of potential applications in functional genomics. Dev. Growth Differ. 2019, 61, 316–326. [Google Scholar] [CrossRef] [Green Version]
- Song, B.; Mott, R.; Gan, X. Recovery of novel association loci in Arabidopsis thaliana and Drosophila melanogaster through leveraging INDELs association and integrated burden test. PLoS Genet. 2018, 14, e1007699. [Google Scholar] [CrossRef] [Green Version]
- Rosen, B.D.; Bickhart, D.M.; Schnabel, R.D.; Koren, S.; Elsik, C.G.; Tseng, E.; Rowan, T.N.; Low, W.Y.; Zimin, A.; Couldrey, C.; et al. De novo assembly of the cattle reference genome with single-molecule sequencing. Gigascience 2020, 9. [Google Scholar] [CrossRef] [Green Version]
- Cretu Stancu, M.; van Roosmalen, M.J.; Renkens, I.; Nieboer, M.M.; Middelkamp, S.; de Ligt, J.; Pregno, G.; Giachino, D.; Mandrile, G.; Espejo Valle-Inclan, J.; et al. Mapping and phasing of structural variation in patient genomes using nanopore sequencing. Nat. Commun. 2017, 8, 1326. [Google Scholar] [CrossRef] [Green Version]
- Norris, A.L.; Workman, R.E.; Fan, Y.; Eshleman, J.R.; Timp, W. Nanopore sequencing detects structural variants in cancer. Cancer Biol. Ther. 2016, 17, 246–253. [Google Scholar] [CrossRef]
- Wray, N.R.; Yang, J.; Hayes, B.J.; Price, A.L.; Goddard, M.E.; Visscher, P.M. Pitfalls of predicting complex traits from SNPs. Nat. Rev. Genet. 2013, 14, 507–515. [Google Scholar] [CrossRef] [Green Version]
- Haile-Mariam, M.; Nieuwhof, G.J.; Beard, K.T.; Konstatinov, K.V.; Hayes, B.J. Comparison of heritabilities of dairy traits in Australian Holstein-Friesian cattle from genomic and pedigree data and implications for genomic evaluations. J. Anim Breed. Genet. 2013, 130, 20–31. [Google Scholar] [CrossRef]
- Theunissen, F.; Flynn, L.L.; Anderton, R.S.; Mastaglia, F.; Pytte, J.; Jiang, L.; Hodgetts, S.; Burns, D.K.; Saunders, A.; Fletcher, S.; et al. Structural Variants May Be a Source of Missing Heritability in sALS. Front. Neurosci-Switz 2020, 14. [Google Scholar] [CrossRef] [Green Version]
- Manolio, T.A.; Collins, F.S.; Cox, N.J.; Goldstein, D.B.; Hindorff, L.A.; Hunter, D.J.; McCarthy, M.I.; Ramos, E.M.; Cardon, L.R.; Chakravarti, A.; et al. Finding the missing heritability of complex diseases. Nature 2009, 461, 747–753. [Google Scholar] [CrossRef] [Green Version]
- Ravenhall, M.; Benavente, E.D.; Sutherland, C.J.; Baker, D.A.; Campino, S.; Clark, T.G. An analysis of large structural variation in global Plasmodium falciparum isolates identifies a novel duplication of the chloroquine resistance associated gene. Sci. Rep. 2019, 9, 8287. [Google Scholar] [CrossRef]
- Rodríguez-Santiago, B.; Malats, N.; Rothman, N.; Armengol, L.; Garcia-Closas, M.; Kogevinas, M.; Villa, O.; Hutchinson, A.; Earl, J.; Marenne, G.; et al. Mosaic uniparental disomies and aneuploidies as large structural variants of the human genome. Am. J. Hum. Genet. 2010, 87, 129–138. [Google Scholar] [CrossRef] [Green Version]
- Walsh, T.; McClellan, J.M.; McCarthy, S.E.; Addington, A.M.; Pierce, S.B.; Cooper, G.M.; Nord, A.S.; Kusenda, M.; Malhotra, D.; Bhandari, A.; et al. Rare Structural Variants Disrupt Multiple Genes in Neurodevelopmental Pathways in Schizophrenia. Science 2008, 320, 539–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harewood, L.; Chaignat, E.; Reymond, A. Structural variation and its effect on expression. Methods Mol. Biol. 2012, 838, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Lupski, J.R.; de Oca-Luna, R.M.; Slaugenhaupt, S.; Pentao, L.; Guzzetta, V.; Trask, B.J.; Saucedo-Cardenas, O.; Barker, D.F.; Killian, J.M.; Garcia, C.A.; et al. DNA duplication associated with Charcot-Marie-Tooth disease type 1A. Cell 1991, 66, 219–232. [Google Scholar] [CrossRef]
- Gonzalez, E.; Kulkarni, H.; Bolivar, H.; Mangano, A.; Sanchez, R.; Catano, G.; Nibbs, R.J.; Freedman, B.I.; Quinones, M.P.; Bamshad, M.J.; et al. The influence of CCL3L1 gene-containing segmental duplications on HIV-1/AIDS susceptibility. Science 2005, 307, 1434–1440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayes, M.; Magano, L.F.; Rivera, N.; Flores, R.; Perez Jurado, L.A. Mutational mechanisms of Williams-Beuren syndrome deletions. Am. J. Hum. Genet. 2003, 73, 131–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bi, W.; Yan, J.; Stankiewicz, P.; Park, S.S.; Walz, K.; Boerkoel, C.F.; Potocki, L.; Shaffer, L.G.; Devriendt, K.; Nowaczyk, M.J.; et al. Genes in a refined Smith-Magenis syndrome critical deletion interval on chromosome 17p11.2 and the syntenic region of the mouse. Genome Res. 2002, 12, 713–728. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Chung, E.K.; Wu, Y.L.; Savelli, S.L.; Nagaraja, H.N.; Zhou, B.; Hebert, M.; Jones, K.N.; Shu, Y.; Kitzmiller, K.; et al. Gene copy-number variation and associated polymorphisms of complement component C4 in human systemic lupus erythematosus (SLE): Low copy number is a risk factor for and high copy number is a protective factor against SLE susceptibility in European Americans. Am. J. Hum. Genet. 2007, 80, 1037–1054. [Google Scholar] [CrossRef] [Green Version]
- Shanta, O.; Noor, A.; Sebat, J.; Chaisson, M.J.P.; Sanders, A.D.; Zhao, X.F.; Malhotra, A.; Porubsky, D.; Rausch, T.; Gardner, E.J.; et al. The effects of common structural variants on 3D chromatin structure. BMC Genom. 2020, 21. [Google Scholar] [CrossRef] [Green Version]
- Bianco, S.; Lupianez, D.G.; Chiariello, A.M.; Annunziatella, C.; Kraft, K.; Schopflin, R.; Wittler, L.; Andrey, G.; Vingron, M.; Pombo, A.; et al. Polymer physics predicts the effects of structural variants on chromatin architecture. Nat. Genet. 2018, 50, 662–667. [Google Scholar] [CrossRef]
- Sanders, S.J.; Ercan-Sencicek, A.G.; Hus, V.; Luo, R.; Murtha, M.T.; Moreno-De-Luca, D.; Chu, S.H.; Moreau, M.P.; Gupta, A.R.; Thomson, S.A.; et al. Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron 2011, 70, 863–885. [Google Scholar] [CrossRef] [Green Version]
- Craddock, N.; Hurles, M.E.; Cardin, N.; Pearson, R.D.; Plagnol, V.; Robson, S.; Vukcevic, D.; Barnes, C.; Conrad, D.F.; Giannoulatou, E.; et al. Genome-wide association study of CNVs in 16,000 cases of eight common diseases and 3,000 shared controls. Nature 2010, 464, 713–U786. [Google Scholar] [CrossRef] [PubMed]
- Williams, N.M.; Zaharieva, I.; Martin, A.; Langley, K.; Mantripragada, K.; Fossdal, R.; Stefansson, H.; Stefansson, K.; Magnusson, P.; Gudmundsson, O.O.; et al. Rare chromosomal deletions and duplications in attention-deficit hyperactivity disorder: A genome-wide analysis. Lancet 2010, 376, 1401–1408. [Google Scholar] [CrossRef] [Green Version]
- Roses, A.D.; Akkari, P.A.; Chiba-Falek, O.; Lutz, M.W.; Gottschalk, W.K.; Saunders, A.M.; Saul, B.; Sundseth, S.; Burns, D. Structural variants can be more informative for disease diagnostics, prognostics and translation than current SNP mapping and exon sequencing. Expert Opin. Drug Met. 2016, 12, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Roses, A.D.; Lutz, M.W.; Amrine-Madsen, H.; Saunders, A.M.; Crenshaw, D.G.; Sundseth, S.S.; Huentelman, M.J.; Welsh-Bohmer, K.A.; Reiman, E.M. A TOMM40 variable-length polymorphism predicts the age of late-onset Alzheimer’s disease. Pharm. J. 2010, 10, 375–384. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, M.; Emerson, J.J.; Macdonald, S.J.; Long, A.D. Structural variants exhibit widespread allelic heterogeneity and shape variation in complex traits. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Voichek, Y.; Weigel, D. Identifying genetic variants underlying phenotypic variation in plants without complete genomes. Nat. Genet. 2020, 52, 534–540. [Google Scholar] [CrossRef]
- Mitsuhashi, S.; Nakagawa, S.; Ueda, M.T.; Imanishi, T.; Frith, M.C.; Mitsuhashi, H. Nanopore-based single molecule sequencing of the D4Z4 array responsible for facioscapulohumeral muscular dystrophy. Sci. Rep. UK 2017, 7. [Google Scholar] [CrossRef] [Green Version]
- Jin, Y.; Cai, H.; Liu, J.; Lin, F.; Qi, X.; Bai, Y.; Lei, C.; Chen, H.; Lan, X. The 10 bp duplication insertion/deletion in the promoter region within paired box 7 gene is associated with growth traits in cattle. Arch. Anim. Breed. 2016, 59, 469–476. [Google Scholar] [CrossRef]
- Mariasegaram, M.; Harrison, B.E.; Bolton, J.A.; Tier, B.; Henshall, J.M.; Barendse, W.; Prayaga, K.C. Fine-mapping the POLL locus in Brahman cattle yields the diagnostic marker CSAFG29. Anim. Genet.. 2012, 43, 683–688. [Google Scholar] [CrossRef]
- Allais-Bonnet, A.; Grohs, C.; Medugorac, I.; Krebs, S.; Djari, A.; Graf, A.; Fritz, S.; Seichter, D.; Baur, A.; Russ, I.; et al. Novel Insights into the Bovine Polled Phenotype and Horn Ontogenesis in Bovidae. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [Green Version]
- Drogemuller, C.; Wohlke, A.; Momke, S.; Distl, O. Fine mapping of the polled locus to a 1-Mb region on bovine chromosome 1q12. Mamm Genome 2005, 16, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Long, C.R.; Gregory, K.E. Inheritance of the Horned, Scurred, and Polled Condition in Cattle. J. Hered 1978, 69, 395–400. [Google Scholar] [CrossRef]
- Crystal Ketel, M.A.-C. Investigating candidate scur genes in Bos taurus breeds. J. Anim. Sci. 2019, 97, 226–227. [Google Scholar] [CrossRef]
- Medugorac, I.; Seichter, D.; Graf, A.; Russ, I.; Blum, H.; Gopel, K.H.; Rothammer, S.; Forster, M.; Krebs, S. Bovine polledness--an autosomal dominant trait with allelic heterogeneity. PLoS ONE 2012, 7, e39477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiedemar, N.; Tetens, J.; Jagannathan, V.; Menoud, A.; Neuenschwander, S.; Bruggmann, R.; Thaller, G.; Drogemuller, C. Independent polled mutations leading to complex gene expression differences in cattle. PLoS ONE 2014, 9, e93435. [Google Scholar] [CrossRef] [Green Version]
- Kent, M.; Moser, M.; Boman, I.A.; Lindtveit, K.; Árnyasi, M.; Sundsaasen, K.K.; Våge, D.I. Insertion of an endogenous Jaagsiekte Sheep Retrovirus element into the BCO2-gene abolishes its function and leads to yellow discoloration of adipose tissue in Norwegian Spælsau (Ovis aries). bioRxiv 2020. [Google Scholar] [CrossRef]
- Durkin, K.; Coppieters, W.; Drögemüller, C.; Ahariz, N.; Cambisano, N.; Druet, T.; Fasquelle, C.; Haile, A.; Horin, P.; Huang, L.; et al. Serial translocation by means of circular intermediates underlies colour sidedness in cattle. Nature 2012, 482, 81–84. [Google Scholar] [CrossRef]
- Awasthi Mishra, N.; Drögemüller, C.; Jagannathan, V.; Keller, I.; Wüthrich, D.; Bruggmann, R.; Beck, J.; Schütz, E.; Brenig, B.; Demmel, S.; et al. A structural variant in the 5’-flanking region of the TWIST2 gene affects melanocyte development in belted cattle. PLoS ONE 2017, 12, e0180170. [Google Scholar] [CrossRef]
- Brenig, B.; Beck, J.; Floren, C.; Bornemann-Kolatzki, K.; Wiedemann, I.; Hennecke, S.; Swalve, H.; Schütz, E. Molecular genetics of coat colour variations in White Galloway and White Park cattle. Anim. Genet. 2013, 44, 450–453. [Google Scholar] [CrossRef]
- Rothammer, S.; Kunz, E.; Krebs, S.; Bitzer, F.; Hauser, A.; Zinovieva, N.; Klymiuk, N.; Medugorac, I. Remapping of the belted phenotype in cattle on BTA3 identifies a multiplication event as the candidate causal mutation. Genet. Sel. Evol. 2018, 50. [Google Scholar] [CrossRef] [Green Version]
- Lamb, H.J.; Ross, E.M.; Nguyen, L.T.; Lyons, R.E.; Moore, S.S.; Hayes, B.J. Characterization of the poll allele in Brahman cattle using long-read Oxford Nanopore sequencing. J. Anim. Sci. 2020, 98. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.Y.; Song, J.; Liu, Y.; Song, C.X.; Yi, C. Mapping the epigenetic modifications of DNA and RNA. Protein Cell 2020, 11, 792–808. [Google Scholar] [CrossRef] [PubMed]
- Leger, A.; Amaral, P.P.; Pandolfini, L.; Capitanchik, C.; Capraro, F.; Barbieri, I.; Migliori, V.; Luscombe, N.M.; Enright, A.J.; Tzelepis, K.; et al. RNA modifications detection by comparative Nanopore direct RNA sequencing. bioRxiv 2019. [Google Scholar] [CrossRef] [Green Version]
- Greenberg, M.V.C.; Bourc’his, D. The diverse roles of DNA methylation in mammalian development and disease. Nat. Rev. Mol. Cell Biol. 2019, 20, 590–607. [Google Scholar] [CrossRef] [PubMed]
- Unnikrishnan, A.; Freeman, W.M.; Jackson, J.; Wren, J.D.; Porter, H.; Richardson, A. The role of DNA methylation in epigenetics of aging. Pharmacy 2019, 195, 172–185. [Google Scholar] [CrossRef]
- Gentilini, D.; Mari, D.; Castaldi, D.; Remondini, D.; Ogliari, G.; Ostan, R.; Bucci, L.; Sirchia, S.M.; Tabano, S.; Cavagnini, F.; et al. Role of epigenetics in human aging and longevity: Genome-wide DNA methylation profile in centenarians and centenarians’ offspring. Age (Dordr) 2013, 35, 1961–1973. [Google Scholar] [CrossRef] [Green Version]
- Adey, A.; Shendure, J. Ultra-low-input, tagmentation-based whole-genome bisulfite sequencing. Genome Res. 2012, 22, 1139–1143. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, S.; Hong, K.; Liu, R.; Shen, L.; Inoue, A.; Diep, D.; Zhang, K.; Zhang, Y. Tet1 controls meiosis by regulating meiotic gene expression. Nature 2012, 492, 443–447. [Google Scholar] [CrossRef] [Green Version]
- Dominissini, D.; Nachtergaele, S.; Moshitch-Moshkovitz, S.; Peer, E.; Kol, N.; Ben-Haim, M.S.; Dai, Q.; Di Segni, A.; Salmon-Divon, M.; Clark, W.C.; et al. The dynamic N(1)-methyladenosine methylome in eukaryotic messenger RNA. Nature 2016, 530, 441–446. [Google Scholar] [CrossRef] [Green Version]
- Marioni, R.E.; Belsky, D.W.; Deary, I.J.; Wagner, W. Association of facial ageing with DNA methylation and epigenetic age predictions. Clin. Epigenet. 2018, 10, 140. [Google Scholar] [CrossRef]
- Tharakan, R.; Ubaida-Mohien, C.; Moore, A.Z.; Hernandez, D.; Tanaka, T.; Ferrucci, L. Blood DNA Methylation and Aging: A Cross-Sectional Analysis and Longitudinal Validation in the InCHIANTI Study. J. Gerontol. A Biol. Sci. Med. Sci. 2020, 75, 2051–2055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barres, R.; Zierath, J.R. DNA methylation in metabolic disorders. Am. J. Clin. Nutr. 2011, 93, 897S–900S. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Kadarmideen, H.N. An Epigenome-Wide DNA Methylation Map of Testis in Pigs for Study of Complex Traits. Front. Genet. 2019, 10, 405. [Google Scholar] [CrossRef] [PubMed]
- McGilchrist, P.; Alston, C.L.; Gardner, G.E.; Thomson, K.L.; Pethick, D.W. Beef carcasses with larger eye muscle areas, lower ossification scores and improved nutrition have a lower incidence of dark cutting. Meat Sci. 2012, 92, 474–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferguson, D.M.; Bruce, H.L.; Thompson, J.M.; Egan, A.F.; Perry, D.; Shorthose, W.R. Factors affecting beef palatability—Farmgate to chilled carcass. Aust. J. Exp. Agric. 2001, 41, 879–891. [Google Scholar] [CrossRef] [Green Version]
- Johnson, A.A.; Akman, K.; Calimport, S.R.; Wuttke, D.; Stolzing, A.; de Magalhaes, J.P. The role of DNA methylation in aging, rejuvenation, and age-related disease. Rejuvenation Res. 2012, 15, 483–494. [Google Scholar] [CrossRef] [Green Version]
- Ciccarone, F.; Tagliatesta, S.; Caiafa, P.; Zampieri, M. DNA methylation dynamics in aging: How far are we from understanding the mechanisms? Mech. Ageing Dev. 2018, 174, 3–17. [Google Scholar] [CrossRef]
- Shinozaki, G.; Braun, P.R.; Hing, B.W.Q.; Ratanatharathorn, A.; Klisares, M.J.; Duncan, G.N.; Jellison, S.S.; Heinzman, J.T.; Nagahama, Y.; Close, L.; et al. Epigenetics of Delirium and Aging: Potential Role of DNA Methylation Change on Cytokine Genes in Glia and Blood Along With Aging. Front. Aging Neurosci. 2018, 10, 311. [Google Scholar] [CrossRef] [Green Version]
- Kosten, T.A.; Huang, W.; Nielsen, D.A. Sex and litter effects on anxiety and DNA methylation levels of stress and neurotrophin genes in adolescent rats. Dev. Psychobiol. 2014, 56, 392–406. [Google Scholar] [CrossRef]
- Klengel, T.; Pape, J.; Binder, E.B.; Mehta, D. The role of DNA methylation in stress-related psychiatric disorders. Neuropharmacology 2014, 80, 115–132. [Google Scholar] [CrossRef]
- Meuwissen, T.H.E.; Hayes, B.J.; Goddard, M.E. Prediction of total genetic value using genome-wide dense marker maps. Genetics 2001, 157, 1819–1829. [Google Scholar] [PubMed]
- Georges, M.; Charlier, C.; Hayes, B. Harnessing genomic information for livestock improvement. Nat. Rev. Genet. 2019, 20, 135–156. [Google Scholar] [CrossRef] [PubMed]
- Ganal, M.W.; Polley, A.; Graner, E.M.; Plieske, J.; Wieseke, R.; Luerssen, H.; Durstewitz, G. Large SNP arrays for genotyping in crop plants. J. Biosci. 2012, 37, 821–828. [Google Scholar] [CrossRef] [PubMed]
- Malmberg, M.M.; Spangenberg, G.C.; Daetwyler, H.D.; Cogan, N.O.I. Assessment of low-coverage nanopore long read sequencing for SNP genotyping in doubled haploid canola (Brassica napus L.). Sci. Rep.-UK 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Ros-Freixedes, R.; Gonen, S.; Gorjanc, G.; Hickey, J.M. A method for allocating low-coverage sequencing resources by targeting haplotypes rather than individuals. Genet. Sel. Evol. 2017, 49. [Google Scholar] [CrossRef] [Green Version]
- Ros-Freixedes, R.; Whalen, A.; Gorjanc, G.; Mileham, A.J.; Hickey, J.M. Evaluation of sequencing strategies for whole-genome imputation with hybrid peeling. Genet. Sel. Evol. 2020, 52, 18. [Google Scholar] [CrossRef] [Green Version]
- Whalen, A.; Ros-Freixedes, R.; Wilson, D.L.; Gorjanc, G.; Hickey, J.M. Hybrid peeling for fast and accurate calling, phasing, and imputation with sequence data of any coverage in pedigrees. Genet. Sel. Evol. 2018, 50, 67. [Google Scholar] [CrossRef] [Green Version]
- Li, J.H.; Mazur, C.A.; Berisa, T.; Pickrell, J.K. Low-pass sequencing increases the power of GWAS and decreases measurement error of polygenic risk scores compared to genotyping arrays. bioRxiv 2020. [Google Scholar] [CrossRef]
- Zou, Y.; Mason, M.G.; Wang, Y.; Wee, E.; Turni, C.; Blackall, P.J.; Trau, M.; Botella, J.R. Correction: Nucleic acid purification from plants, animals and microbes in under 30 seconds. PLoS Biol. 2018, 16, e1002630. [Google Scholar] [CrossRef]
- Xu, Y.; Yang-Turner, F.; Volk, D.; Crook, D. NanoSPC: A scalable, portable, cloud compatible viral nanopore metagenomic data processing pipeline. Nucleic Acids Res. 2020, 48, W366–W371. [Google Scholar] [CrossRef]
- Grumaz, C.; Hoffmann, A.; Vainshtein, Y.; Kopp, M.; Grumaz, S.; Stevens, P.; Decker, S.O.; Weigand, M.A.; Hofer, S.; Brenner, T.; et al. Rapid Next-Generation Sequencing-Based Diagnostics of Bacteremia in Septic Patients. J. Mol. Diagn. 2020, 22, 405–418. [Google Scholar] [CrossRef] [Green Version]
- Boykin, L.M.; Sseruwagi, P.; Alicai, T.; Ateka, E.; Mohammed, I.U.; Stanton, J.A.L.; Kayuki, C.; Mark, D.; Fute, T.; Erasto, J.; et al. Tree Lab: Portable Genomics for Early Detection of Plant Viruses and Pests in Sub-Saharan Africa. Genes-Basel 2019, 10, 632. [Google Scholar] [CrossRef] [Green Version]
- Sanderson, N.D.; Street, T.L.; Foster, D.; Swann, J.; Atkins, B.L.; Brent, A.J.; McNally, M.A.; Oakley, S.; Taylor, A.; Peto, T.E.A.; et al. Real-time analysis of nanopore-based metagenomic sequencing from infected orthopaedic devices. BMC Genom. 2018, 19, 714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wick, R.R.; Judd, L.M.; Holt, K.E. Performance of neural network basecalling tools for Oxford Nanopore sequencing. Genome Biol. 2019, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deshpande, S.V.; Reed, T.M.; Sullivan, R.E.; Kerkhof, L.J.; Beigel, K.M.; Wade, M.M. Offline Next Generation Metagenomics Sequence Analysis Using MinION Detection Software (MINDS). Genes-Basel 2019, 10, 578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Copley Pastoral Company. Crows Nest; Copley Pastoral Company: Winya, QLD, Australia, 2020. [Google Scholar]
- Schaeffer, L.R. Strategy for applying genome-wide selection in dairy cattle. J. Anim. Breed. Genet. 2006, 123, 218–223. [Google Scholar] [CrossRef]
- Peñagaricano, F. Chapter 6—Genetics and genomics of dairy cattle. In Animal Agriculture; Bazer, F.W., Lamb, G.C., Wu, G., Eds.; Academic Press: Cambridge, MA, USA, 2020; pp. 101–119. [Google Scholar]
- Mikheyev, A.S.; Tin, M.M.Y. A first look at the Oxford Nanopore MinION sequencer. Mol. Ecol. Resour. 2014, 14, 1097–1102. [Google Scholar] [CrossRef]
- Laver, T.; Harrison, J.; O’Neill, P.A.; Moore, K.; Farbos, A.; Paszkiewicz, K.; Studholme, D.J. Assessing the performance of the Oxford Nanopore Technologies MinION. Biomol. Detect. Quantif 2015, 3, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Zeng, J.; Cai, H.; Peng, H.; Wang, H.; Zhang, Y.; Akutsu, T. Causalcall: Nanopore Basecalling Using a Temporal Convolutional Network. Front. Genet. 2020, 10. [Google Scholar] [CrossRef] [Green Version]
- Gilpatrick, T.; Lee, I.; Graham, J.E.; Raimondeau, E.; Bowen, R.; Heron, A.; Sedlazeck, F.J.; Timp, W. Targeted Nanopore Sequencing with Cas9 for studies of methylation, structural variants and mutations. bioRxiv 2019. [Google Scholar] [CrossRef] [Green Version]
- López-Girona, E.; Davy, M.W.; Albert, N.W.; Hilario, E.; Smart, M.E.M.; Kirk, C.; Thomson, S.J.; Chagné, D. CRISPR-Cas9 enrichment and long read sequencing for fine mapping in plants. Plant Methods 2020, 16, 121. [Google Scholar] [CrossRef] [PubMed]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A Robust, Simple Genotyping-by-Sequencing (GBS) Approach for High Diversity Species. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romay, M.C.; Millard, M.J.; Glaubitz, J.C.; Peiffer, J.A.; Swarts, K.L.; Casstevens, T.M.; Elshire, R.J.; Acharya, C.B.; Mitchell, S.E.; Flint-Garcia, S.A.; et al. Comprehensive genotyping of the USA national maize inbred seed bank. Genome Biol. 2013, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crossa, J.; Beyene, Y.; Kassa, S.; Perez, P.; Hickey, J.M.; Chen, C.; de los Campos, G.; Burgueno, J.; Windhausen, V.S.; Buckler, E.; et al. Genomic Prediction in Maize Breeding Populations with Genotyping-by-Sequencing. G3-Genes Genom Genet. 2013, 3, 1903–1926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodds, K.G.; McEwan, J.C.; Brauning, R.; Anderson, R.M.; van Stijn, T.C.; Kristjansson, T.; Clarke, S.M. Construction of relatedness matrices using genotyping-by-sequencing data. BMC Genom. 2015, 16. [Google Scholar] [CrossRef] [Green Version]
- Yue, J.X.; Liti, G.N. simuG: A general-purpose genome simulator. Bioinformatics 2019, 35, 4442–4444. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Han, R.; Bi, C.; Li, M.; Wang, S.; Gao, X. DeepSimulator: A deep simulator for Nanopore sequencing. Bioinformatics 2018, 34, 2899–2908. [Google Scholar] [CrossRef] [Green Version]
- De Coster, W.; D’Hert, S.; Schultz, D.T.; Cruts, M.; Van Broeckhoven, C. NanoPack: Visualizing and processing long-read sequencing data. Bioinformatics 2018, 34, 2666–2669. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Lund, M.S.; Ma, P.; Janss, L.; Kadarmideen, H.N.; Su, G. Improving genomic predictions by correction of genotypes from genotyping by sequencing in livestock populations. J. Anim. Sci. Biotechnol. 2019, 10, 8. [Google Scholar] [CrossRef] [PubMed]
- Maruki, T.; Lynch, M. Genotype Calling from Population-Genomic Sequencing Data. G3: Genes|Genomes|Genet. 2017, 7, 1393–1404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, E.R.; Kinnamon, D.D.; Schmidt, M.A.; Powell, E.H.; Zuchner, S.; Morris, R.W. SeqEM: An adaptive genotype-calling approach for next-generation sequencing studies. Bioinformatics 2010, 26, 2803–2810. [Google Scholar] [CrossRef]
- Dassonneville, R.; Brondum, R.F.; Druet, T.; Fritz, S.; Guillaume, F.; Guldbrandtsen, B.; Lund, M.S.; Ducrocq, V.; Su, G. Effect of imputing markers from a low-density chip on the reliability of genomic breeding values in Holstein populations. J. Dairy Sci 2011, 94, 3679–3686. [Google Scholar] [CrossRef] [Green Version]
- Howie, B.N.; Donnelly, P.; Marchini, J. A Flexible and Accurate Genotype Imputation Method for the Next Generation of Genome-Wide Association Studies. PLoS Genet. 2009, 5. [Google Scholar] [CrossRef] [Green Version]
- Mulder, H.A.; Calus, M.P.L.; Druet, T.; Schrooten, C. Imputation of genotypes with low-density chips and its effect on reliability of direct genomic values in Dutch Holstein cattle. J. Dairy Sci. 2012, 95, 876–889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dassonneville, R.; Fritz, S.; Ducrocq, V.; Boichard, D. Short communication: Imputation performances of 3 low-density marker panels in beef and dairy cattle. J. Dairy Sci 2012, 95, 4136–4140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrooten, C.; Dassonneville, R.; Ducrocq, V.; Brondum, R.F.; Lund, M.S.; Chen, J.; Liu, Z.T.; Gonzalez-Recio, O.; Pena, J.; Druet, T. Error rate for imputation from the Illumina BovineSNP50 chip to the Illumina BovineHD chip. Genet. Sel. Evol. 2014, 46. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lamb, H.J.; Hayes, B.J.; Nguyen, L.T.; Ross, E.M. The Future of Livestock Management: A Review of Real-Time Portable Sequencing Applied to Livestock. Genes 2020, 11, 1478. https://doi.org/10.3390/genes11121478
Lamb HJ, Hayes BJ, Nguyen LT, Ross EM. The Future of Livestock Management: A Review of Real-Time Portable Sequencing Applied to Livestock. Genes. 2020; 11(12):1478. https://doi.org/10.3390/genes11121478
Chicago/Turabian StyleLamb, Harrison J., Ben J. Hayes, Loan T. Nguyen, and Elizabeth M. Ross. 2020. "The Future of Livestock Management: A Review of Real-Time Portable Sequencing Applied to Livestock" Genes 11, no. 12: 1478. https://doi.org/10.3390/genes11121478
APA StyleLamb, H. J., Hayes, B. J., Nguyen, L. T., & Ross, E. M. (2020). The Future of Livestock Management: A Review of Real-Time Portable Sequencing Applied to Livestock. Genes, 11(12), 1478. https://doi.org/10.3390/genes11121478