RAM-PGK: Prediction of Lysine Phosphoglycerylation Based on Residue Adjacency Matrix


 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Protein Dataset
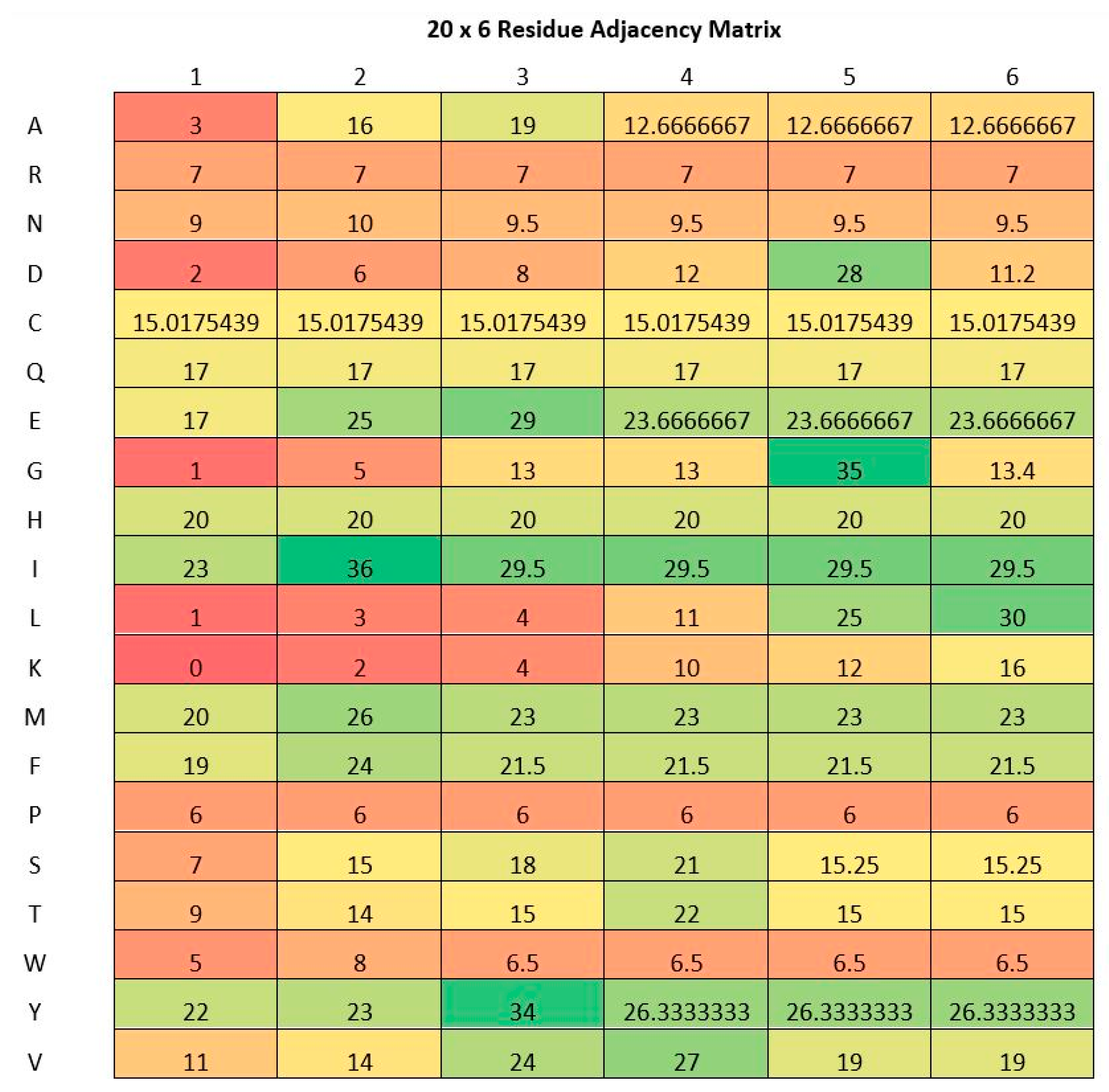
2.2. Residue Adjacency Matrix
2.3. Support Vector Machine
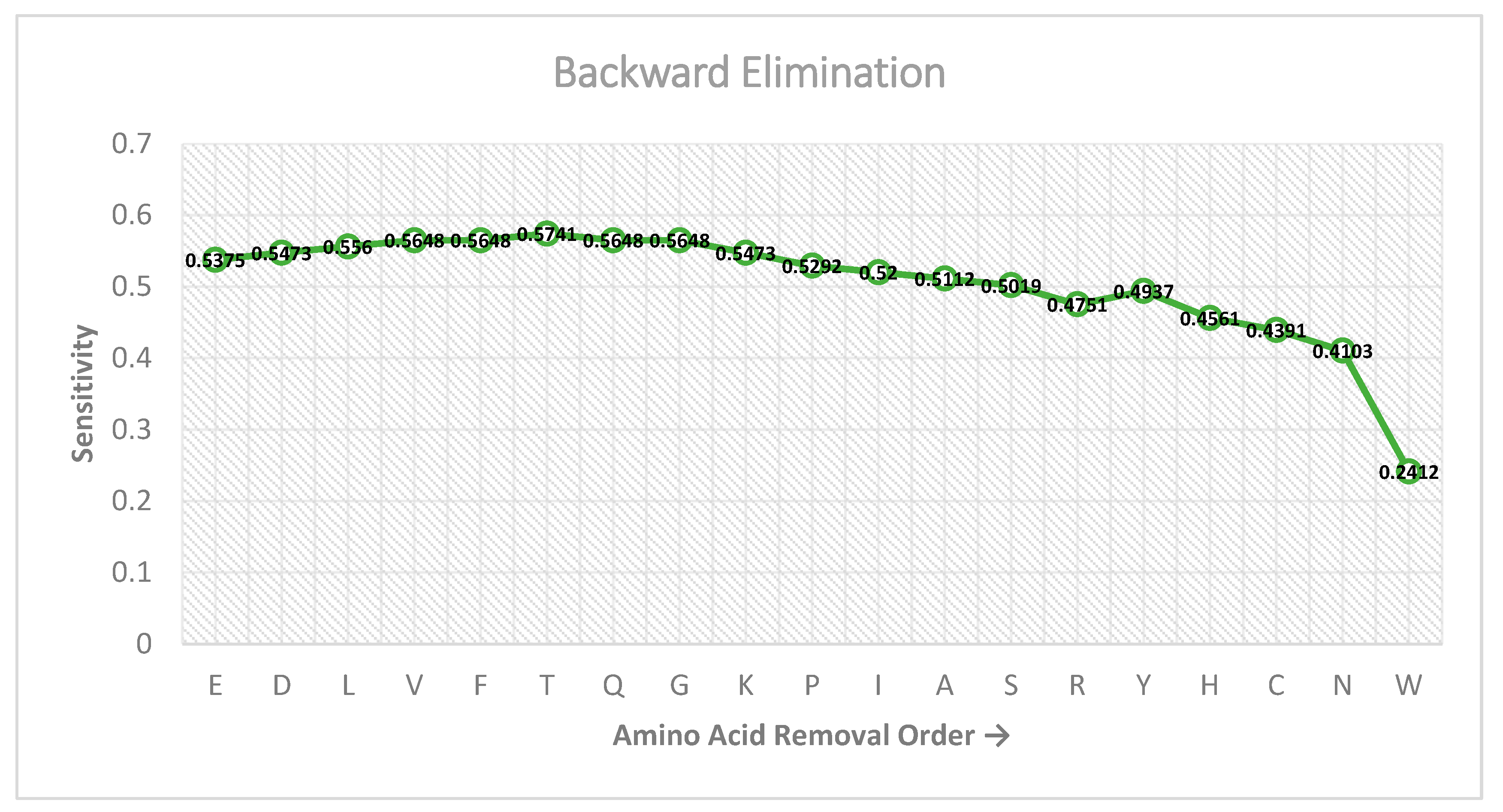
2.4. Feature Selection Scheme
3. Results and Discussion
3.1. Dataset Balancing
3.2. Statistical Measures
3.3. Test Scheme
- Step 1: Divide the dataset into six similar parts.
- Step 2: Combine the five parts and apply the cleaning treatment to balance the positive and negative classes. Train the predictor using this balanced dataset and test it with the part left out.
- Step 3: Set the predictor parameters with the train set.
- Step 4: Acquire the statistical measures on the test set.
- Step 5: Repeat steps 2 to 4 for the remainder of the folds.
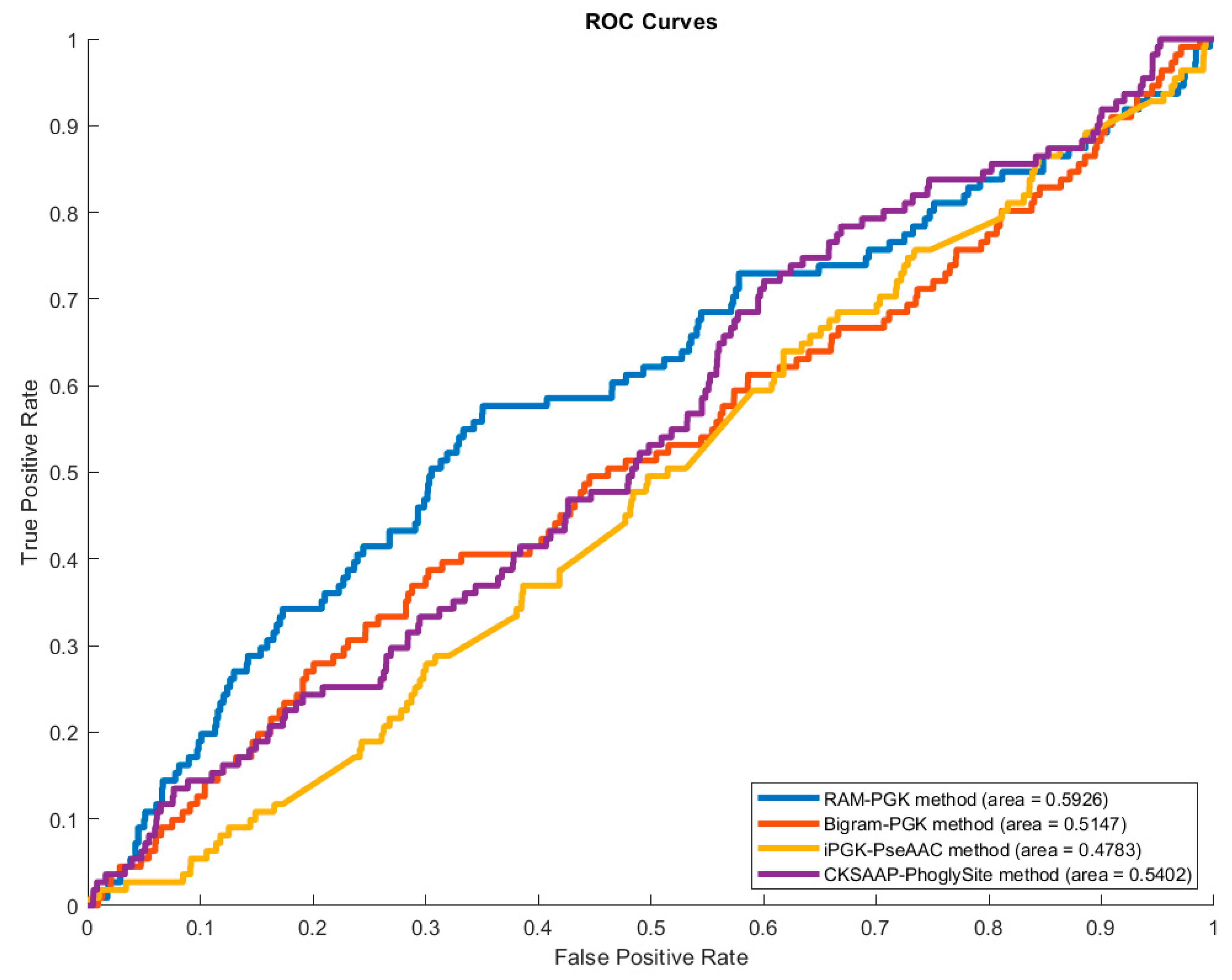
3.4. Comparison of RAM-PGK with iPGK-PseAAC Predictor
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| PTM | Post-translational modification |
| RAM | Residue adjacency matrix |
| MCC | Mathews correlation coefficient |
| SVM | Support vector machine |
| PLMD | Protein Lysine Modification Database |
References
- Huang, J.; Wang, F.; Ye, M.; Zou, H. Enrichment and separation techniques for large-scale proteomics analysis of the protein post-translational modifications. J. Chromatogr. A 2014, 1372, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Lanouette, S.; Mongeon, V.; Figeys, D.; Couture, J.F. The functional diversity of protein lysine methylation. Mol. Syst. Biol. 2014, 10, 724. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Wang, Y.; Gao, T.; Pan, Z.; Cheng, H.; Yang, Q.; Cheng, Z.; Guo, A.; Ren, J.; Xue, Y. CPLM: A database of protein lysine modifications. Nucleic Acids Res. 2014, 42, D531–D536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, K.-C. An unprecedented revolution in medicinal chemistry driven by the progress of biological science. Curr. Top. Med. Chem. 2017, 17, 2337–2358. [Google Scholar] [CrossRef] [PubMed]
- Lan, F.; Shi, Y. Epigenetic regulation: Methylation of histone and non-histone proteins. Sci. China Ser. C Life Sci. 2009, 52, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Iyer, L.M.; Burroughs, A.M.; Aravind, L. Unraveling the biochemistry and provenance of pupylation: A prokaryotic analog of ubiquitination. Biol. Direct 2008, 3, 45. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Chen, Y.; Tishkoff, D.X.; Peng, C.; Tan, M.; Dai, L.; Xie, Z.; Zhang, Y.; Zwaans, B.M.; Skinner, M.E. SIRT5-mediated lysine desuccinylation impacts diverse metabolic pathways. Mol. Cell 2013, 50, 919–930. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Z.; Tang, Y.; Chen, Y.; Kim, S.; Liu, H.; Li, S.S.; Gu, W.; Zhao, Y. Molecular characterization of propionyllysines in non-histone proteins. Mol. Cell. Proteom. 2009, 8, 45–52. [Google Scholar] [CrossRef] [Green Version]
- Tan, M.; Luo, H.; Lee, S.; Jin, F.; Yang, J.S.; Montellier, E.; Buchou, T.; Cheng, Z.; Rousseaux, S.; Rajagopal, N. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell 2011, 146, 1016–1028. [Google Scholar] [CrossRef] [Green Version]
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009, 325, 834–840. [Google Scholar] [CrossRef] [Green Version]
- Reddy, H.M.; Sharma, A.; Dehzangi, A.; Shigemizu, D.; Chandra, A.A.; Tsunoda, T. GlyStruct: Glycation prediction using structural properties of amino acid residues. BMC Bioinform. 2019, 19, 547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansen, M.B.; Kiemer, L.; Brunak, S. Analysis and prediction of mammalian protein glycation. Glycobiology 2006, 16, 844–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szondy, Z.; Korponay-Szabó, I.; Király, R.; Sarang, Z.; Tsay, G.J. Transglutaminase 2 in human diseases. BioMedicine 2017, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Iakoucheva, L.M.; Mooney, S.D.; Radivojac, P. Loss of post-translational modification sites in disease. In Biocomputing 2010; World Scientific: Kamuela, HI, USA, 2010; pp. 337–347. [Google Scholar]
- Liddy, K.A.; White, M.Y.; Cordwell, S.J. Functional decorations: Post-translational modifications and heart disease delineated by targeted proteomics. Genome Med. 2013, 5, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spinelli, F.R.; Pecani, A.; Conti, F.; Mancini, R.; Alessandri, C.; Valesini, G. Post-translational modifications in rheumatoid arthritis and atherosclerosis: Focus on citrullination and carbamylation. J. Int. Med. Res. 2016, 44, 81–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ju, Z.; Cao, J.-Z.; Gu, H. Predicting lysine phosphoglycerylation with fuzzy SVM by incorporating k-spaced amino acid pairs into Chou’s general PseAAC. J. Theor. Biol. 2016, 397, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Moellering, R.E.; Cravatt, B.F. Functional lysine modification by an intrinsically reactive primary glycolytic metabolite. Science 2013, 341, 549–553. [Google Scholar] [CrossRef] [Green Version]
- Bulcun, E.; Ekici, M.; Ekici, A. Disorders of glucose metabolism and insulin resistance in patients with obstructive sleep apnoea syndrome. Int. J. Clin. Pract. 2012, 66, 91–97. [Google Scholar] [CrossRef]
- Kolwicz, S.C., Jr.; Tian, R. Glucose metabolism and cardiac hypertrophy. Cardiovasc. Res. 2011, 90, 194–201. [Google Scholar] [CrossRef] [Green Version]
- López, Y.; Sharma, A.; Dehzangi, A.; Lal, S.P.; Taherzadeh, G.; Sattar, A.; Tsunoda, T. Success: Evolutionary and structural properties of amino acids prove effective for succinylation site prediction. BMC Genom. 2018, 19, 923. [Google Scholar] [CrossRef]
- Ju, Z.; He, J.-J. Prediction of lysine propionylation sites using biased SVM and incorporating four different sequence features into Chou’s PseAAC. J. Mol. Graph. Model. 2017, 76, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Ding, Y.-X.; Ding, J.; Wu, L.-Y.; Xue, Y. Mal-Lys: Prediction of lysine malonylation sites in proteins integrated sequence-based features with mRMR feature selection. Sci. Rep. 2016, 6, 38318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, Q.; Feng, K.; Liao, B.; Liu, Y.; Huang, G. Prediction of Lysine Malonylation Sites Based on Pseudo Amino Acid. Comb. Chem. High Throughput Screen. 2017, 20, 622–628. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Zhai, Z.; Li, Y.; Lu, M.; Cai, T.; Zhou, B.; Huang, L.; Wei, T.; Li, T. Prediction of Protein Lysine Acylation by Integrating Primary Sequence Information with Multiple Functional Features. J. Proteome Res. 2016, 15, 4234–4244. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.-R.; Xiao, X.; Lin, W.-Z.; Chou, K.-C. iUbiq-Lys: Prediction of lysine ubiquitination sites in proteins by extracting sequence evolution information via a gray system model. J. Biomol. Struct. Dyn. 2015, 33, 1731–1742. [Google Scholar] [CrossRef]
- Hou, T.; Zheng, G.; Zhang, P.; Jia, J.; Li, J.; Xie, L.; Wei, C.; Li, Y. LAceP: Lysine acetylation site prediction using logistic regression classifiers. PLoS ONE 2014, 9, e89575. [Google Scholar] [CrossRef] [Green Version]
- Jia, J.; Zhang, L.; Liu, Z.; Xiao, X.; Chou, K.-C. pSumo-CD: Predicting sumoylation sites in proteins with covariance discriminant algorithm by incorporating sequence-coupled effects into general PseAAC. Bioinformatics 2016, 32, 3133–3141. [Google Scholar] [CrossRef] [Green Version]
- Qiu, W.-R.; Sun, B.-Q.; Xiao, X.; Xu, Z.-C.; Jia, J.-H.; Chou, K.-C. iKcr-PseEns: Identify lysine crotonylation sites in histone proteins with pseudo components and ensemble classifier. Genomics 2018, 110, 239–246. [Google Scholar] [CrossRef]
- Ju, Z.; Gu, H. Predicting pupylation sites in prokaryotic proteins using semi-supervised self-training support vector machine algorithm. Anal. Biochem. 2016, 507, 1–6. [Google Scholar] [CrossRef]
- Bakhtiarizadeh, M.R.; Moradi-Shahrbabak, M.; Ebrahimi, M.; Ebrahimie, E. Neural network and SVM classifiers accurately predict lipid binding proteins, irrespective of sequence homology. J. Theor. Biol. 2014, 356, 213–222. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, M.; Xi, J.; Luo, F.; Li, A. PTM-ssMP: A Web Server for Predicting Different Types of Post-translational Modification Sites Using Novel Site-specific Modification Profile. Int. J. Biol. Sci. 2018, 14, 946–956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Wang, M.; Li, A. Prediction of post-translational modification sites using multiple kernel support vector machine. PeerJ 2017, 5, e3261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, W.; Xu, X.; Shen, Y.; Feng, H.; Li, A.; Wang, M. Prediction of protein kinase-specific phosphorylation sites in hierarchical structure using functional information and random forest. Amino Acids 2014, 46, 1069–1078. [Google Scholar] [CrossRef] [PubMed]
- Chandra, A.; Sharma, A.; Dehzangi, A.; Ranganathan, S.; Jokhan, A.; Chou, K.-C.; Tsunoda, T. PhoglyStruct: Prediction of phosphoglycerylated lysine residues using structural properties of amino acids. Sci. Rep. 2018, 8, 17923. [Google Scholar] [CrossRef] [PubMed]
- Dehzangi, A.; López, Y.; Lal, S.P.; Taherzadeh, G.; Michaelson, J.; Sattar, A.; Tsunoda, T.; Sharma, A. PSSM-Suc: Accurately predicting succinylation using position specific scoring matrix into bigram for feature extraction. J. Theor. Biol. 2017, 425, 97–102. [Google Scholar] [CrossRef]
- Chou, K.-C.; Shen, H.-B. Recent progress in protein subcellular location prediction. Anal. Biochem. 2007, 370, 1–16. [Google Scholar] [CrossRef]
- Jia, J.; Liu, Z.; Xiao, X.; Liu, B.; Chou, K.-C. iSuc-PseOpt: Identifying lysine succinylation sites in proteins by incorporating sequence-coupling effects into pseudo components and optimizing imbalanced training dataset. Anal. Biochem. 2016, 497, 48–56. [Google Scholar] [CrossRef]
- Xu, Y.; Ding, Y.-X.; Ding, J.; Wu, L.-Y.; Deng, N.-Y. Phogly–PseAAC: Prediction of lysine phosphoglycerylation in proteins incorporating with position-specific propensity. J. Theor. Biol. 2015, 379, 10–15. [Google Scholar] [CrossRef]
- Chen, Q.-Y.; Tang, J.; Du, P.-F. Predicting protein lysine phosphoglycerylation sites by hybridizing many sequence based features. Mol. Biosyst. 2017, 13, 874–882. [Google Scholar] [CrossRef]
- Chandra, A.A.; Sharma, A.; Dehzangi, A.; Tsunoda, T. EvolStruct-Phogly: Incorporating structural properties and evolutionary information from profile bigrams for the phosphoglycerylation prediction. BMC Genom. 2019, 19, 984. [Google Scholar] [CrossRef] [Green Version]
- Chandra, A.; Sharma, A.; Dehzangi, A.; Shigemizu, D.; Tsunoda, T. Bigram-PGK: Phosphoglycerylation prediction using the technique of bigram probabilities of position specific scoring matrix. BMC Mol. Cell Biol. 2019, 20, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.-M.; Xu, Y.; Chou, K.-C. iPGK-PseAAC: Identify lysine phosphoglycerylation sites in proteins by incorporating four different tiers of amino acid pairwise coupling information into the general PseAAC. Med. Chem. 2017, 13, 552–559. [Google Scholar] [CrossRef] [PubMed]
- Dehzangi, A.; López, Y.; Lal, S.P.; Taherzadeh, G.; Sattar, A.; Tsunoda, T.; Sharma, A. Improving succinylation prediction accuracy by incorporating the secondary structure via helix, strand and coil, and evolutionary information from profile bigrams. PLoS ONE 2018, 13, e0191900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López, Y.; Dehzangi, A.; Lal, S.P.; Taherzadeh, G.; Michaelson, J.; Sattar, A.; Tsunoda, T.; Sharma, A. SucStruct: Prediction of succinylated lysine residues by using structural properties of amino acids. Anal. Biochem. 2017, 527, 24–32. [Google Scholar] [CrossRef]
- Mapes, N.J., Jr.; Rodriguez, C.; Chowriappa, P.; Dua, S. Residue adjacency matrix based feature engineering for predicting cysteine reactivity in proteins. Comput. Struct. Biotechnol. J. 2019, 17, 90–100. [Google Scholar] [CrossRef]
- Sharma, A.; Paliwal, K.K.; Dehzangi, A.; Lyons, J.; Imoto, S.; Miyano, S. A strategy to select suitable physicochemical attributes of amino acids for protein fold recognition. BMC Bioinform. 2013, 14, 233. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Xiao, X.; Qiu, W.-R.; Chou, K.-C. iDNA-Methyl: Identifying DNA methylation sites via pseudo trinucleotide composition. Anal. Biochem. 2015, 474, 69–77. [Google Scholar] [CrossRef]
- Chen, W.; Feng, P.; Ding, H.; Lin, H.; Chou, K.-C. iRNA-Methyl: Identifying N6-methyladenosine sites using pseudo nucleotide composition. Anal. Biochem. 2015, 490, 26–33. [Google Scholar] [CrossRef]
- Liu, B.; Fang, L.; Wang, S.; Wang, X.; Li, H.; Chou, K.-C. Identification of microRNA precursor with the degenerate K-tuple or Kmer strategy. J. Theor. Biol. 2015, 385, 153–159. [Google Scholar] [CrossRef]
- Ding, H.; Deng, E.-Z.; Yuan, L.-F.; Liu, L.; Lin, H.; Chen, W.; Chou, K.-C. iCTX-Type: A sequence-based predictor for identifying the types of conotoxins in targeting ion channels. BioMed Res. Int. 2014. [Google Scholar] [CrossRef]
- Xiao, X.; Min, J.-L.; Lin, W.-Z.; Liu, Z.; Cheng, X.; Chou, K.-C. iDrug-Target: Predicting the interactions between drug compounds and target proteins in cellular networking via benchmark dataset optimization approach. J. Biomol. Struct. Dyn. 2015, 33, 2221–2233. [Google Scholar] [CrossRef] [PubMed]
- Chou, K.-C.; Zhang, C.-T. Prediction of protein structural classes. Crit. Rev. Biochem. Mol. Biol. 1995, 30, 275–349. [Google Scholar] [CrossRef] [PubMed]
- Chou, K.C. Prediction of protein cellular attributes using pseudo-amino acid composition. Proteins Struct. Funct. Bioinform. 2001, 43, 246–255. [Google Scholar] [CrossRef] [PubMed]
- Hajisharifi, Z.; Piryaiee, M.; Beigi, M.M.; Behbahani, M.; Mohabatkar, H. Predicting anticancer peptides with Chou’s pseudo amino acid composition and investigating their mutagenicity via Ames test. J. Theor. Biol. 2014, 341, 34–40. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| 1 | 2 | 3 | |
| A | 1 | 1 | 3 |
| K | 0 | 2 | N/A |
| M | 2 | N/A | N/A |
| Y | N/A | N/A | N/A |
| A | 1 | 1 | 3 |
| K | 0 | 2 | 1 |
| M | 2 | 2 | 2 |
| Y | 1.5 | 1.5 | 1.5 |
| Predictor | Sensitivity | Specificity | Precision | Accuracy | MCC |
|---|---|---|---|---|---|
| CKSAAP_PhoglySite [17] | 0.3494 | 0.6722 | 0.0358 | 0.6616 | 0.0090 |
| iPGK-PseAAC [43] | 0.0185 | 0.9791 | 0.0064 | 0.9473 | −0.5048 |
| Bigram-PGK [42] | 0.4055 | 0.6639 | 0.0428 | 0.6554 | 0.0292 |
| RAM-PGK (No Feature Selection) | 0.5380 | 0.6328 | 0.0472 | 0.6298 | 0.0631 |
| RAM-PGK | 0.5741 | 0.6436 | 0.0531 | 0.6414 | 0.0824 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chandra, A.A.; Sharma, A.; Dehzangi, A.; Tsunoda, T. RAM-PGK: Prediction of Lysine Phosphoglycerylation Based on Residue Adjacency Matrix. Genes 2020, 11, 1524. https://doi.org/10.3390/genes11121524
Chandra AA, Sharma A, Dehzangi A, Tsunoda T. RAM-PGK: Prediction of Lysine Phosphoglycerylation Based on Residue Adjacency Matrix. Genes. 2020; 11(12):1524. https://doi.org/10.3390/genes11121524
Chicago/Turabian StyleChandra, Abel Avitesh, Alok Sharma, Abdollah Dehzangi, and Tatushiko Tsunoda. 2020. "RAM-PGK: Prediction of Lysine Phosphoglycerylation Based on Residue Adjacency Matrix" Genes 11, no. 12: 1524. https://doi.org/10.3390/genes11121524
APA StyleChandra, A. A., Sharma, A., Dehzangi, A., & Tsunoda, T. (2020). RAM-PGK: Prediction of Lysine Phosphoglycerylation Based on Residue Adjacency Matrix. Genes, 11(12), 1524. https://doi.org/10.3390/genes11121524