Reconstituting the Mammalian Apoptotic Switch in Yeast

Abstract
:
{kind=link}
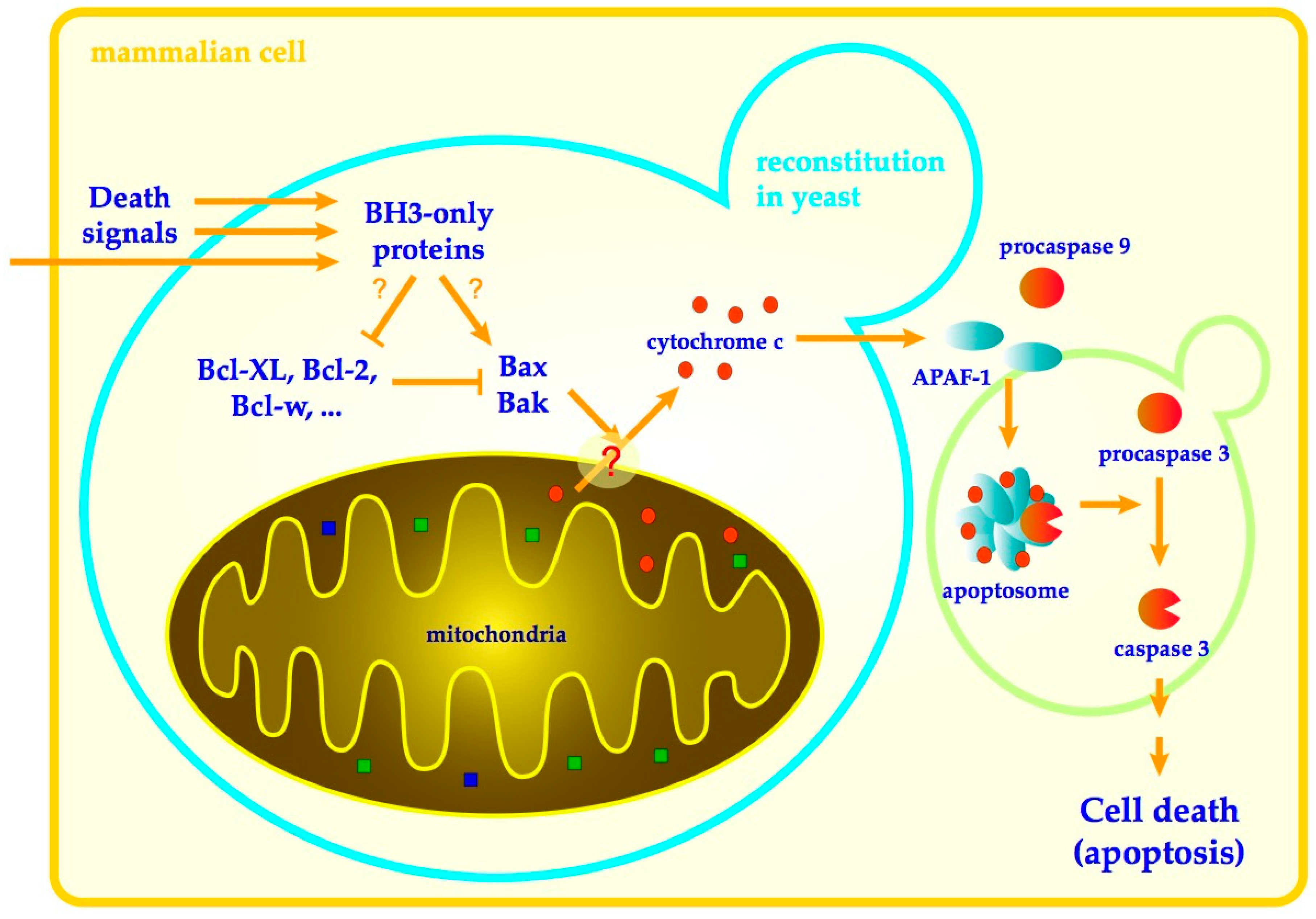{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Bcl-2 Proteins Expressed in Yeast
3. Bax and Bak Activation
4. Inhibition of Bax and Bak by Antiapoptotic Proteins
5. BH3-only Proteins
6. Anticancer Drugs
7. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Adams, J.M.; Cory, S. The Bcl-2 apoptotic switch in cancer development and therapy. Oncogene 2007, 26, 1324–1337. [Google Scholar] [CrossRef] [Green Version]
- Green, D.R.; Fitzgerald, P. Just So Stories about the Evolution of Apoptosis. Curr. Biol. 2016, 26, R620–R627. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Kass, G.E.N.; Szegezdi, E.; Joseph, B. The mitochondrial death pathway: A promising therapeutic target in diseases. J. Cell Mol. Med. 2009, 13, 1004–1033. [Google Scholar] [CrossRef] [Green Version]
- Bratton, S.B.; Salvesen, G.S. Regulation of the Apaf-1-caspase-9 apoptosome. J. Cell Sci. 2010, 123, 3209–3214. [Google Scholar] [CrossRef] [Green Version]
- Hawkins, C.J.; Silke, J.; Verhagen, A.M.; Foster, R.; Ekert, P.G.; Ashley, D.M. Analysis of candidate antagonists of IAP-mediated caspase inhibition using yeast reconstituted with the mammalian Apaf-1-activated apoptosis mechanism. Apoptosis 2001, 6, 331–338. [Google Scholar] [CrossRef]
- Kvansakul, M.; Hinds, M.G. The Structural Biology of BH3-Only Proteins. Regul. Cell Death Pt A: Apoptotic Mech. 2014, 544, 49–74. [Google Scholar]
- Bakhshi, A.; Jensen, J.P.; Goldman, P.; Wright, J.J.; McBride, O.W.; Epstein, A.L.; Korsmeyer, S.J. Cloning the Chromosomal Breakpoint of T(14-18) Human Lymphomas—Clustering around Jh on Chromosome-14 and near a Transcriptional Unit on 18. Cell 1985, 41, 899–906. [Google Scholar] [CrossRef]
- Cleary, M.L.; Sklar, J. Nucleotide-Sequence of a T(14,18) Chromosomal Breakpoint in Follicular Lymphoma and Demonstration of a Breakpoint-Cluster Region near a Transcriptionally Active Locus on Chromosome-18. Proc. Natl. Acad. Sci. USA 1985, 82, 7439–7443. [Google Scholar] [CrossRef] [Green Version]
- Tsujimoto, Y.; Cossman, J.; Jaffe, E.; Croce, C.M. Involvement of the Bcl-2 Gene in Human Follicular Lymphoma. Science 1985, 228, 1440–1443. [Google Scholar] [CrossRef]
- Youle, R.J.; Strasser, A. The BCL-2 protein family: Opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 2008, 9, 47–59. [Google Scholar] [CrossRef]
- Aouacheria, A.; Rech de Laval, V.; Combet, C.; Hardwick, J.M. Evolution of Bcl-2 homology motifs: Homology versus homoplasy. Trends Cell. Biol. 2012, 23, 103–111. [Google Scholar] [CrossRef]
- Oltvai, Z.N.; Milliman, C.L.; Korsmeyer, S.J. Bcl-2 Heterodimerizes in-Vivo with a Conserved Homolog, Bax, That Accelerates Programmed Cell-Death. Cell 1993, 74, 609–619. [Google Scholar] [CrossRef]
- Chittenden, T.; Harrington, E.A.; Oconnor, R.; Flemington, C.; Lutz, R.J.; Evan, G.I.; Guild, B.C. Induction of Apoptosis by the Bcl-2 Homolog Bak. Nature 1995, 374, 733–736. [Google Scholar] [CrossRef]
- Hsu, S.Y.; Kaipia, A.; McGee, E.; Lomeli, M.; Hsueh, A.J.W. Bok is a pro-apoptotic Bcl-2 protein with restricted expression in reproductive tissues and heterodimerizes with selective anti-apoptotic Bcl-2 family members. Proc. Natl. Acad. Sci. USA 1997, 94, 12401–12406. [Google Scholar] [CrossRef] [Green Version]
- Hsu, Y.T.; Wolter, K.G.; Youle, R.J. Cytosol-to-membrane redistribution of Bax and Bcl-X-L during apoptosis. Proc. Natl. Acad. Sci. USA 1997, 94, 3668–3672. [Google Scholar] [CrossRef] [Green Version]
- Wolter, K.G.; Hsu, Y.T.; Smith, C.L.; Nechushtan, A.; Xi, X.G.; Youle, R.J. Movement of Bax from the cytosol to mitochondria during apoptosis. J. Cell Biol. 1997, 139, 1281–1292. [Google Scholar] [CrossRef]
- Llambi, F.; Wang, Y.M.; Victor, B.; Yang, M.; Schneider, D.M.; Gingras, S.; Parsons, M.J.; Zheng, J.H.; Brown, S.A.; Pelletier, S.; et al. BOK Is a Non-canonical BCL-2 Family Effector of Apoptosis Regulated by ER-Associated Degradation. Cell 2016, 165, 421–433. [Google Scholar] [CrossRef] [Green Version]
- Kaufmann, T.; Schlipf, S.; Sanz, J.; Neubert, K.; Stein, R.; Borner, C. Characterization of the signal that directs Bcl-x(L), but not Bcl-2, to the mitochondrial outer membrane. J. Cell Biol. 2003, 160, 53–64. [Google Scholar] [CrossRef] [Green Version]
- Shibue, T.; Taniguchi, T. BH3-only proteins: Integrated control point of apoptosis. Int. J. Cancer 2006, 119, 2036–2043. [Google Scholar] [CrossRef]
- Happo, L.; Strasser, A.; Cory, S. BH3-only proteins in apoptosis at a glance. J. Cell Sci. 2012, 125, 1081–1087. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, M.; Youle, R.J.; Tjandra, N. Structure of Bax: coregulation of dimer formation and intracellular localization. Cell 2000, 103, 645–654. [Google Scholar] [CrossRef] [Green Version]
- Muchmore, S.W.; Sattler, M.; Liang, H.; Meadows, R.P.; Harlan, J.E.; Yoon, H.S.; Nettesheim, D.; Chang, B.S.; Thompson, C.B.; Wong, S.L.; et al. X-ray and NMR structure of human Bcl-xL, an inhibitor of programmed cell death. Nature 1996, 381, 335–341. [Google Scholar] [CrossRef]
- Petros, A.M.; Olejniczak, E.T.; Fesik, S.W. Structural biology of the Bcl-2 family of proteins. Biochim. Biophys. Acta 2004, 1644, 83–94. [Google Scholar] [CrossRef]
- Rautureau, G.J.; Day, C.L.; Hinds, M.G. Intrinsically disordered proteins in bcl-2 regulated apoptosis. Int. J. Mol. Sci. 2010, 11, 1808–1824. [Google Scholar] [CrossRef] [Green Version]
- Carmona-Gutierrez, D.; Eisenberg, T.; Buttner, S.; Meisinger, C.; Kroemer, G.; Madeo, F. Apoptosis in yeast: Triggers, pathways, subroutines. Cell Death Differ. 2010, 17, 763–773. [Google Scholar] [CrossRef] [Green Version]
- Carmona-Gutierrez, D.; Bauer, M.A.; Zimmermann, A.; Aguilera, A.; Austriaco, N.; Ayscough, K.; Balzan, R.; Bar-Nun, S.; Barrientos, A.; Belenky, P.; et al. Guidelines and recommendations on yeast cell death nomenclature. Microb. Cell 2018, 5, 4–31. [Google Scholar] [CrossRef] [Green Version]
- Sato, T.; Hanada, M.; Bodrug, S.; Irie, S.; Iwama, N.; Boise, L.H.; Thompson, C.B.; Golemis, E.; Fong, L.; Wang, H.G. Interactions among members of the Bcl-2 protein family analyzed with a yeast two-hybrid system. Proc. Natl. Acad. Sci. USA 1994, 91, 9238–9242. [Google Scholar] [CrossRef] [Green Version]
- Zha, H.; Fisk, H.A.; Yaffe, M.P.; Mahajan, N.; Herman, B.; Reed, J.C. Structure-function comparisons of the proapoptotic protein Bax in yeast and mammalian cells. Mol. Cell Biol. 1996, 16, 6494–6508. [Google Scholar] [CrossRef]
- Manon, S.; Chaudhuri, B.; Guerin, M. Release of cytochrome c and decrease of cytochrome c oxidase in Bax-expressing yeast cells, and prevention of these effects by coexpression of Bcl-xL. FEBS Lett. 1997, 415, 29–32. [Google Scholar] [CrossRef] [Green Version]
- Pavlov, E.V.; Priault, M.; Pietkiewicz, D.; Cheng, E.H.; Antonsson, B.; Manon, S.; Korsmeyer, S.J.; Mannella, C.A.; Kinnally, K.W. A novel, high conductance channel of mitochondria linked to apoptosis in mammalian cells and Bax expression in yeast. J. Cell Biol. 2001, 155, 725–731. [Google Scholar] [CrossRef] [Green Version]
- Danial, N.N.; Korsmeyer, S.J. Cell death: Critical control points. Cell 2004, 116, 205–219. [Google Scholar]
- Priault, M.; Camougrand, N.; Chaudhuri, B.; Schaeffer, J.; Manon, S. Comparison of the effects of bax-expression in yeast under fermentative and respiratory conditions: Investigation of the role of adenine nucleotides carrier and cytochrome c. FEBS Lett. 1999, 456, 232–238. [Google Scholar] [CrossRef] [Green Version]
- Gross, A.; Pilcher, K.; Blachly-Dyson, E.; Basso, E.; Jockel, J.; Bassik, M.C.; Korsmeyer, S.J.; Forte, M. Biochemical and genetic analysis of the mitochondrial response of yeast to BAX and BCL-X(L). Mol. Cell Biol. 2000, 20, 3125–3136. [Google Scholar] [CrossRef] [Green Version]
- Kiššová, I.; Polčic, P.; Kempná, P.; Zeman, I.; Šabová, L.; Kolarov, J. The cytotoxic action of Bax on yeast cells does not require mitochondrial ADP/ATP carrier but may be related to its import to the mitochondria. FEBS Lett. 2000, 471, 113–118. [Google Scholar] [CrossRef] [Green Version]
- Pfanner, N. Mitochondrial import: Crossing the aqueous intermembrane space. Curr. Biol. 1998, 8, R262–R265. [Google Scholar] [CrossRef] [Green Version]
- Sanjuan Szklarz, L.K.; Kozjak-Pavlovic, V.; Vogtle, F.N.; Chacinska, A.; Milenkovic, D.; Vogel, S.; Durr, M.; Westermann, B.; Guiard, B.; Martinou, J.C.; et al. Preprotein transport machineries of yeast mitochondrial outer membrane are not required for Bax-induced release of intermembrane space proteins. J. Mol. Biol. 2007, 368, 44–54. [Google Scholar] [CrossRef] [Green Version]
- Ligr, M.; Madeo, F.; Frohlich, E.; Hilt, W.; Frohlich, K.U.; Wolf, D.H. Mammalian Bax triggers apoptotic changes in yeast. FEBS Lett. 1998, 438, 61–65. [Google Scholar] [CrossRef] [Green Version]
- Polčic, P.; Jaká, P.; Mentel, M. Yeast as a tool for studying proteins of the Bcl-2 family. Microb. Cell 2015, 2, 74–87. [Google Scholar] [CrossRef] [Green Version]
- Legiot, A.; Cere, C.; Dupoiron, T.; Kaabouni, M.; Camougrand, N.; Manon, S. Mitochondria-Associated Membranes (MAMs) are involved in Bax mitochondria! localization and cytochrome c release. Microb. Cell 2019, 6, 257–266. [Google Scholar] [CrossRef]
- Herrera-Cruz, M.S.; Simmen, T. Over Six Decades of Discovery and Characterization of the Architecture at Mitochondria-Associated Membranes (MAMs). In Organelle Contact Sites: From Molecular Mechanism to Disease; Springer: Singapore, 2017; Volume 997, pp. 13–31. [Google Scholar] [CrossRef]
- Willis, S.N.; Adams, J.M. Life in the balance: How BH3-only proteins induce apoptosis. Curr. Opin. Cell Biol. 2005, 17, 617–625. [Google Scholar] [CrossRef] [Green Version]
- Renault, T.T.; Manon, S. Bax: Addressed to kill. Biochimie 2011, 93, 1379–1391. [Google Scholar] [CrossRef]
- Greenhalf, W.; Stephan, C.; Chaudhuri, B. Role of mitochondria and C-terminal membrane anchor of Bcl-2 in Bax induced growth arrest and mortality in Saccharomyces cerevisiae. FEBS Lett. 1996, 380, 169–175. [Google Scholar] [CrossRef] [Green Version]
- Tao, W.; Kurschner, C.; Morgan, J.I. Modulation of cell death in yeast by the Bcl-2 family of proteins. J. Biol. Chem. 1997, 272, 15547–15552. [Google Scholar] [CrossRef] [Green Version]
- Matsuyama, S.; Xu, Q.; Velours, J.; Reed, J.C. The Mitochondrial F0F1-ATPase proton pump is required for function of the proapoptotic protein Bax in yeast and mammalian cells. Mol. Cell 1998, 1, 327–336. [Google Scholar] [CrossRef]
- Xu, Q.; Reed, J.C. Bax inhibitor-1, a mammalian apoptosis suppressor identified by functional screening in yeast. Mol. Cell 1998, 1, 337–346. [Google Scholar] [CrossRef]
- Marzo, I.; Brenner, C.; Zamzami, N.; Jurgensmeier, J.M.; Susin, S.A.; Vieira, H.L.; Prevost, M.C.; Xie, Z.; Matsuyama, S.; Reed, J.C.; et al. Bax and adenine nucleotide translocator cooperate in the mitochondrial control of apoptosis. Science 1998, 281, 2027–2031. [Google Scholar] [CrossRef]
- O’Neill, K.L.; Huang, K.; Zhang, J.J.; Chen, Y.; Luo, X. Inactivation of prosurvival Bcl-2 proteins activates Bax/Bak through the outer mitochondrial membrane. Genes Dev. 2016, 30, 973–988. [Google Scholar] [CrossRef] [Green Version]
- Huang, K.; O’Neill, K.L.; Li, J.; Zhou, W.; Han, N.; Pang, X.; Wu, W.; Struble, L.; Borgstahl, G.; Liu, Z.; et al. BH3-only proteins target BCL-xL/MCL-1, not BAX/BAK, to initiate apoptosis. Cell Res. 2019, 29, 942–952. [Google Scholar] [CrossRef]
- Priault, M.; Cartron, P.F.; Camougrand, N.; Antonsson, B.; Vallette, F.M.; Manon, S. Investigation of the role of the C-terminus of Bax and of tc-Bid on Bax interaction with yeast mitochondria. Cell Death Differ. 2003, 10, 1068–1077. [Google Scholar] [CrossRef] [Green Version]
- Cartron, P.F.; Priault, M.; Oliver, L.; Meflah, K.; Manon, S.; Vallette, F.M. The N-terminal end of Bax contains a mitochondrial-targeting signal. J. Biol. Chem. 2003, 278, 11633–11641. [Google Scholar] [CrossRef] [Green Version]
- Arokium, H.; Camougrand, N.; Vallette, F.M.; Manon, S. Studies of the interaction of substituted mutants of BAX with yeast mitochondria reveal that the C-terminal hydrophobic alpha-helix is a second ART sequence and plays a role in the interaction with anti-apoptotic BCL-xL. J. Biol. Chem. 2004, 279, 52566–52573. [Google Scholar] [CrossRef] [Green Version]
- Simonyan, L.; Legiot, A.; Lascu, I.; Durand, G.; Giraud, M.F.; Gonzalez, C.; Manon, S. The substitution of Proline 168 favors Bax oligomerization and stimulates its interaction with LUVs and mitochondria. Biochim. Biophys. Acta 2017, 1859, 1144–1155. [Google Scholar] [CrossRef]
- Alves, S.; Neiri, L.; Chaves, S.R.; Vieira, S.; Trindade, D.; Manon, S.; Dominguez, V.; Pintado, B.; Jonckheere, V.; Van Damme, P.; et al. N-terminal acetylation modulates Bax targeting to mitochondria. Int. J. Biochem. Cell Biol. 2018, 95, 35–42. [Google Scholar] [CrossRef]
- Banadyga, L.; Lam, S.C.; Okamoto, T.; Kvansakul, M.; Huang, D.C.; Barry, M. Deerpox Virus Encodes an Inhibitor of Apoptosis That Regulates Bak and Bax. J. Virol. 2011, 85, 1922–1934. [Google Scholar] [CrossRef] [Green Version]
- Juhásová, B.; Bhatia-Kiššová, I.; Polčicová, K.; Mentel, M.; Forte, M.; Polčic, P. Reconstitution of interactions of Murine gammaherpesvirus 68 M11 with Bcl-2 family proteins in yeast. Bioch. Biophys. Res. Commun. 2011, 407, 783–787. [Google Scholar] [CrossRef]
- Bloomer, D.T.; Kitevska, T.; Brand, I.L.; Jabbour, A.M.; Nguyen, H.; Hawkins, C.J. Modeling Metazoan Apoptotic Pathways in Yeast. Methods Mol. Biol. 2016, 1419, 161–183. [Google Scholar] [CrossRef]
- Polčic, P.; Forte, M. Response of yeast to the regulated expression of proteins in the Bcl-2 family. Biochem. J. 2003, 374, 393–402. [Google Scholar] [CrossRef] [Green Version]
- Yin, X.M.; Oltvai, Z.N.; Korsmeyer, S.J. BH1 and BH2 domains of Bcl-2 are required for inhibition of apoptosis and heterodimerization with Bax. Nature 1994, 369, 321–323. [Google Scholar] [CrossRef]
- Sedlak, T.W.; Oltvai, Z.N.; Yang, E.; Wang, K.; Boise, L.H.; Thompson, C.B.; Korsmeyer, S.J. Multiple Bcl-2 family members demonstrate selective dimerizations with Bax. Proc. Natl. Acad. Sci. USA 1995, 92, 7834–7838. [Google Scholar] [CrossRef] [Green Version]
- Minn, A.J.; Kettlun, C.S.; Liang, H.; Kelekar, A.; Vander Heiden, M.G.; Chang, B.S.; Fesik, S.W.; Fill, M.; Thompson, C.B. Bcl-xL regulates apoptosis by heterodimerization-dependent and -independent mechanisms. EMBO J. 1999, 18, 632–643. [Google Scholar] [CrossRef] [Green Version]
- Edlich, F.; Banerjee, S.; Suzuki, M.; Cleland, M.M.; Arnoult, D.; Wang, C.; Neutzner, A.; Tjandra, N.; Youle, R.J. Bcl-x(L) retrotranslocates Bax from the mitochondria into the cytosol. Cell 2011, 145, 104–116. [Google Scholar] [CrossRef] [Green Version]
- Renault, T.T.; Teijido, O.; Missire, F.; Ganesan, Y.T.; Velours, G.; Arokium, H.; Beaumatin, F.; Llanos, R.; Athane, A.; Camougrand, N.; et al. Bcl-xL stimulates Bax relocation to mitochondria and primes cells to ABT-737. Int. J. Biochem. Cell Biol. 2015, 64, 136–146. [Google Scholar] [CrossRef]
- Renault, T.T.; Dejean, L.M.; Manon, S. A brewing understanding of the regulation of Bax function by Bcl-xL and Bcl-2. Mech. Ageing Dev. 2017, 161, 201–210. [Google Scholar] [CrossRef]
- Gérecová, G.; Kopanicová, J.; Jaká, P.; Běhalová, L.; Juhásová, B.; Bhatia-Kiššová, I.; Forte, M.; Polčic, P.; Mentel, M. BH3-only proteins Noxa, Bik, Bmf, and Bid activate Bax and Bak indirectly when studied in yeast model. FEMS Yeast Res. 2013, 13, 747–754. [Google Scholar] [CrossRef] [Green Version]
- Juhásová, B.; Mentel, M.; Bhatia-Kiššová, I.; Zeman, I.; Kolarov, J.; Forte, M.; Polčic, P. BH3-only protein Bim inhibits activity of antiapoptotic members of Bcl-2 family when expressed in yeast. FEBS Lett. 2011, 585, 2709–2713. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Yin, X.M.; Chao, D.T.; Milliman, C.L.; Korsmeyer, S.J. BID: A novel BH3 domain-only death agonist. Genes Dev. 1996, 10, 2859–2869. [Google Scholar]
- Luo, X.; Budihardjo, I.; Zou, H.; Slaughter, C.; Wang, X.D. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell 1998, 94, 481–490. [Google Scholar]
- Gross, A.; Yin, X.M.; Wang, K.; Wei, M.C.; Jockel, J.; Millman, C.; Erdjument-Bromage, H.; Tempst, P.; Korsmeyer, S.J. Caspase cleaved BID targets mitochondria and is required for cytochrome c release, while BCL-X-L prevents this release but not tumor necrosis factor-R1/Fas death. J. Biol. Chem. 1999, 274, 1156–1163. [Google Scholar]
- Guscetti, F.; Nath, N.; Denko, N. Functional characterization of human proapoptotic molecules in yeast S. cerevisiae. FASEB J. 2005, 19, 464–466. [Google Scholar] [CrossRef]
- Weber, A.; Paschen, S.A.; Heger, K.; Wilfling, F.; Frankenberg, T.; Bauerschmitt, H.; Seiffert, B.M.; Kirschnek, S.; Wagner, H.; Hacker, G. BimS-induced apoptosis requires mitochondrial localization but not interaction with anti-apoptotic Bcl-2 proteins. J. Cell Biol. 2007, 177, 625–636. [Google Scholar] [CrossRef] [Green Version]
- Wilfling, F.; Weber, A.; Potthoff, S.; Vogtle, F.N.; Meisinger, C.; Paschen, S.A.; Hacker, G. BH3-only proteins are tail-anchored in the outer mitochondrial membrane and can initiate the activation of Bax. Cell Death Differ. 2012, 19, 1328–1336. [Google Scholar] [CrossRef] [Green Version]
- Gallenne, T.; Gautier, F.; Oliver, L.; Hervouet, E.; Noel, B.; Hickman, J.A.; Geneste, O.; Cartron, P.F.; Vallette, F.M.; Manon, S.; et al. Bax activation by the BH3-only protein Puma promotes cell dependence on antiapoptotic Bcl-2 family members. J. Cell Biol. 2009, 185, 279–290. [Google Scholar] [CrossRef] [Green Version]
- Gavathiotis, E.; Suzuki, M.; Davis, M.L.; Pitter, K.; Bird, G.H.; Katz, S.G.; Tu, H.C.; Kim, H.; Cheng, E.H.Y.; Tjandra, N.; et al. BAX activation is initiated at a novel interaction site. Nature 2008, 455, 1076–1081. [Google Scholar] [CrossRef] [Green Version]
- Walensky, L.D.; Pitter, K.; Morash, J.; Oh, K.J.; Barbuto, S.; Fisher, J.; Smith, E.; Verdine, G.L.; Korsmeyer, S.J. A stapled BID BH3 helix directly binds and activates BAX. Mol. Cell 2006, 24, 199–210. [Google Scholar] [CrossRef]
- Buttner, S.; Ruli, D.; Vogtle, F.N.; Galluzzi, L.; Moitzi, B.; Eisenberg, T.; Kepp, O.; Habernig, L.; Carmona-Gutierrez, D.; Rockenfeller, P.; et al. A yeast BH3-only protein mediates the mitochondrial pathway of apoptosis. EMBO J. 2011, 30, 2779–2792. [Google Scholar] [CrossRef] [Green Version]
- Cebulski, J.; Malouin, J.; Pinches, N.; Cascio, V.; Austriaco, N. Yeast Bax inhibitor, Bxi1p, is an ER-localized protein that links the unfolded protein response and programmed cell death in Saccharomyces cerevisiae. PLoS ONE 2011, 6, e20882. [Google Scholar] [CrossRef]
- Kelly, P.N.; Strasser, A. The role of Bcl-2 and its pro-survival relatives in tumourigenesis and cancer therapy. Cell Death Differ. 2011, 18, 1414–1424. [Google Scholar] [CrossRef] [Green Version]
- Merino, D.; Kelly, G.L.; Lessene, G.; Wei, A.H.; Roberts, A.W.; Strasser, A. BH3-Mimetic Drugs: Blazing the Trail for New Cancer Medicines. Cancer Cell 2018, 34, 879–891. [Google Scholar]
- Green, D.R. A BH3 Mimetic for Killing Cancer Cells. Cell 2016, 165, 1560. [Google Scholar] [CrossRef]
- Beaumont, T.E.; Shekhar, T.M.; Kaur, L.; Pantaki-Eimany, D.; Kvansakul, M.; Hawkins, C.J. Yeast techniques for modeling drugs targeting Bcl-2 and caspase family members. Cell Death Dis. 2013, 4, e619. [Google Scholar] [CrossRef] [Green Version]
- Zhai, D.; Jin, C.; Satterthwait, A.C.; Reed, J.C. Comparison of chemical inhibitors of antiapoptotic Bcl-2-family proteins. Cell Death Differ. 2006, 13, 1419–1421. [Google Scholar] [CrossRef] [Green Version]
- Doshi, J.M.; Tian, D.F.; Xing, C.G. Ethyl-2-amino-6-bromo-4-(1-cyano-2-ethoxy-2-oxoethyl)-4H-chromene-3-carboxylate (HA 14-1), a prototype small-molecule antagonist against antiapoptotic bcl-2 proteins, decomposes to generate reactive oxygen species that induce apoptosis. Mol. Pharm. 2007, 4, 919–928. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Polčic, P.; Mentel, M. Reconstituting the Mammalian Apoptotic Switch in Yeast. Genes 2020, 11, 145. https://doi.org/10.3390/genes11020145
Polčic P, Mentel M. Reconstituting the Mammalian Apoptotic Switch in Yeast. Genes. 2020; 11(2):145. https://doi.org/10.3390/genes11020145
Chicago/Turabian StylePolčic, Peter, and Marek Mentel. 2020. "Reconstituting the Mammalian Apoptotic Switch in Yeast" Genes 11, no. 2: 145. https://doi.org/10.3390/genes11020145
APA StylePolčic, P., & Mentel, M. (2020). Reconstituting the Mammalian Apoptotic Switch in Yeast. Genes, 11(2), 145. https://doi.org/10.3390/genes11020145