Comparative Genomics and Evolutionary Analysis of RNA-Binding Proteins of Burkholderia cenocepacia J2315 and Other Members of the B. cepacia Complex



Abstract
:1. Introduction
2. Materials and Methods
2.1. Database Searches for Putative RBPs in B. cenocepacia J2315 Genome
2.2. Putative RBPs in Bcc and Non-Bcc Burkhoderia Genomes
2.3. Multiple Sequence Alignments and Phylogenetic Analyses
2.4. Evolutionary Analysis
2.5. Statistical Analysis
3. Results and Discussion
3.1. Putative “Conventional” RBPs Identified in the B. cenocepacia J2315 Genome
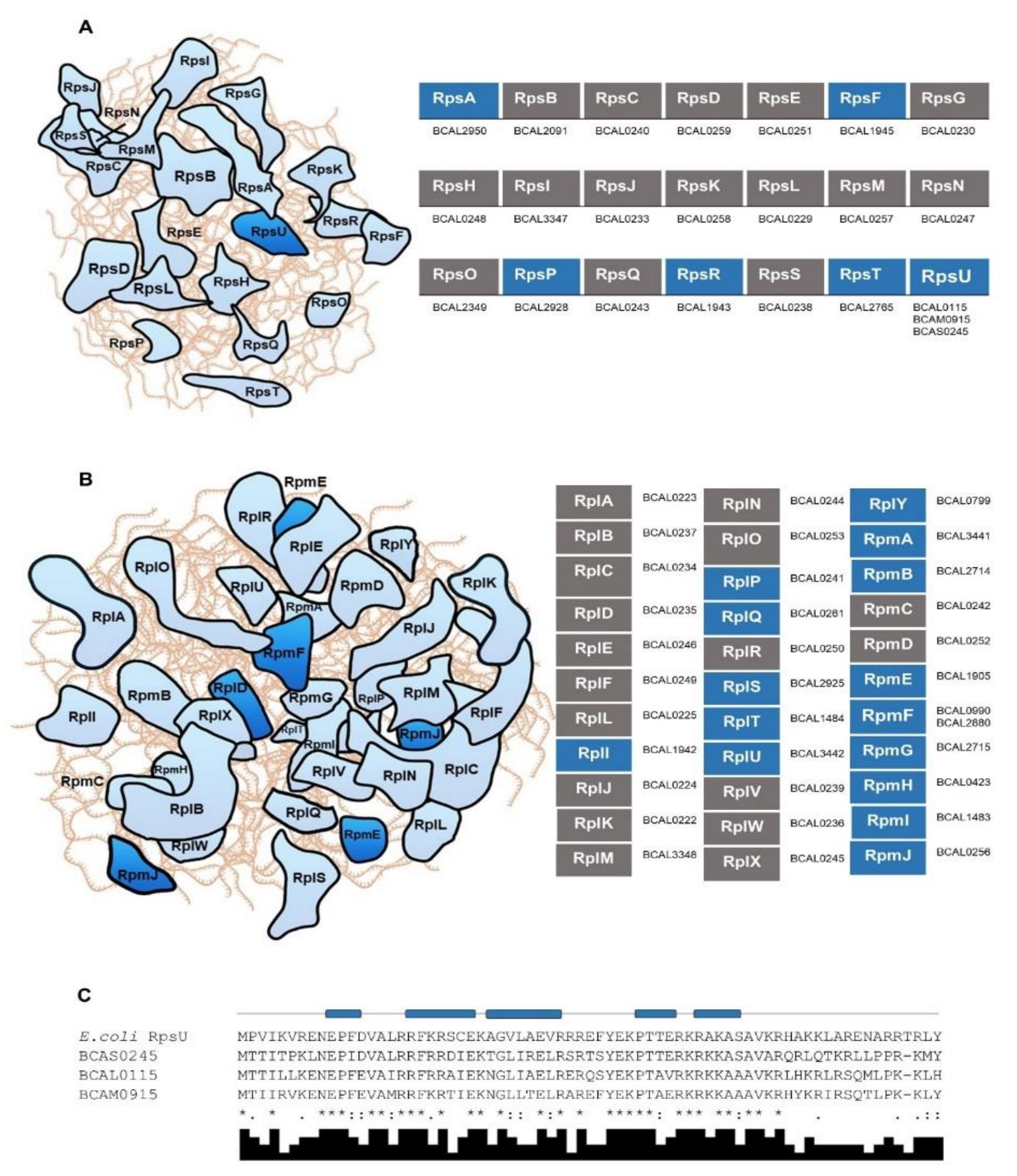
3.2. R-proteins and Other RBPs Involved in Protein Synthesis in B. cenocepacia J2315 and in Other Bacteria of the Burkholderia Genus
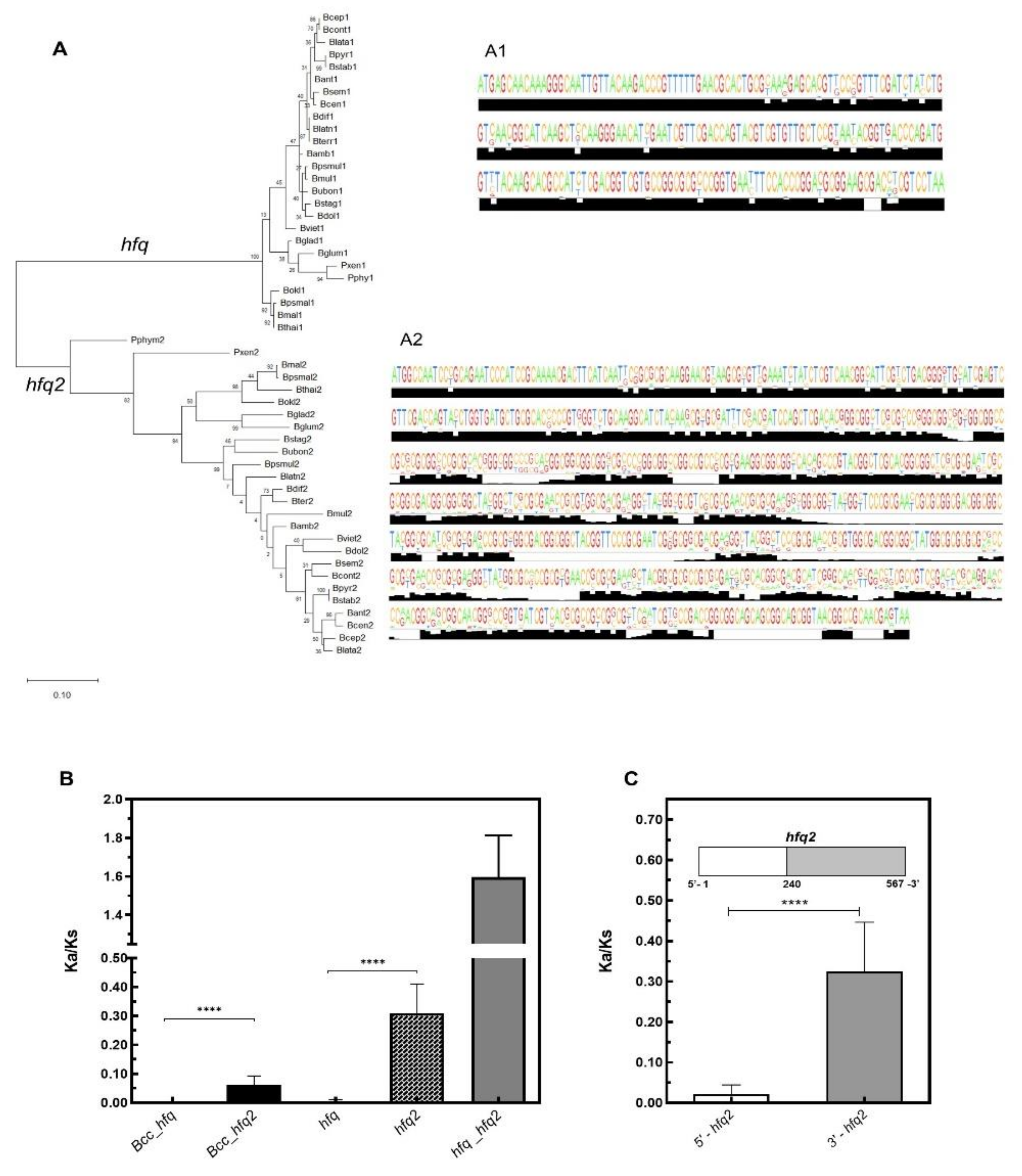
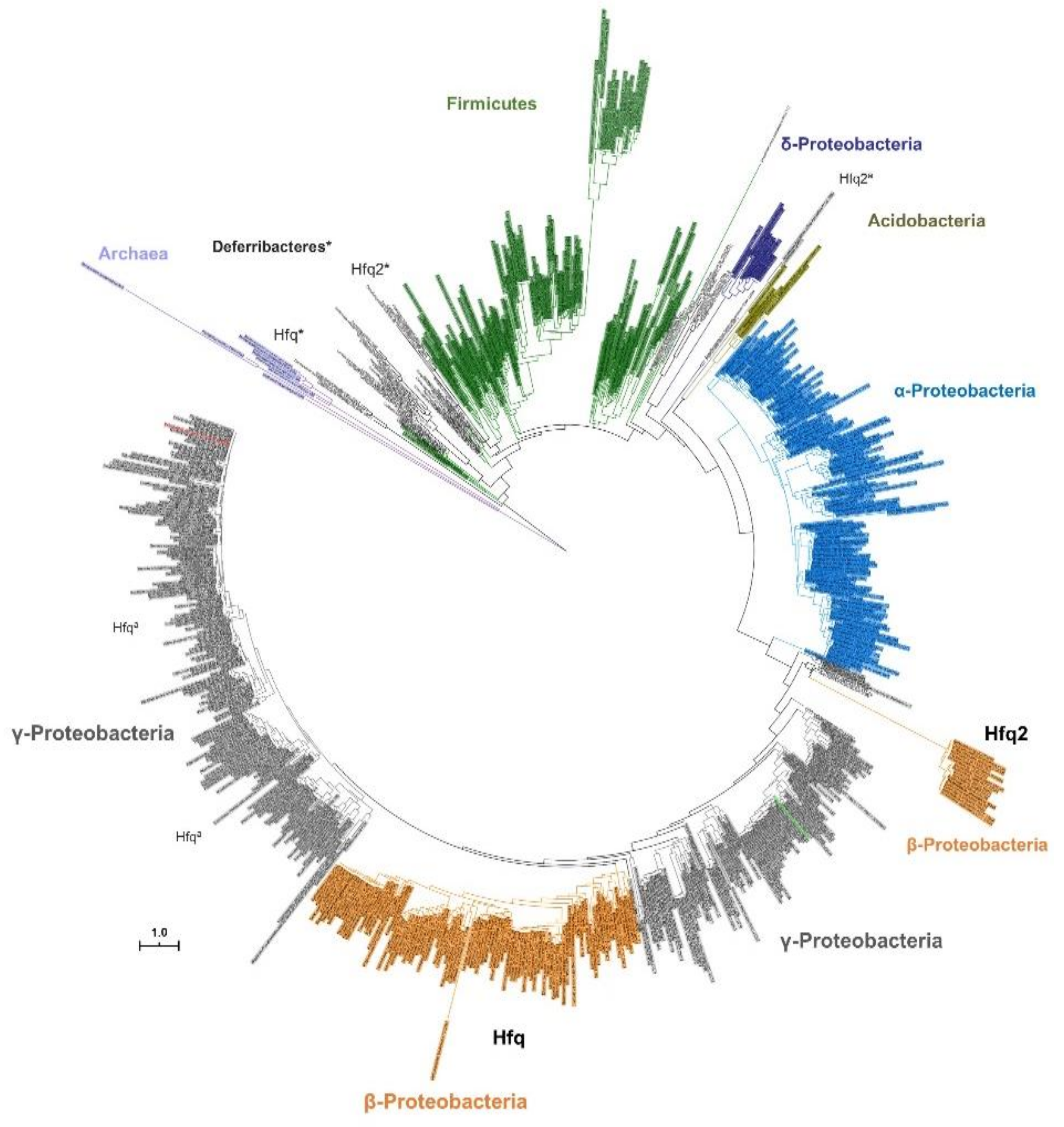
3.3. Contrasting With Hfq2, Hfq is Highly Conserved Among Bcc and is an Ortholog of Hfq Proteins From Other β- and γ-Proteobacteria
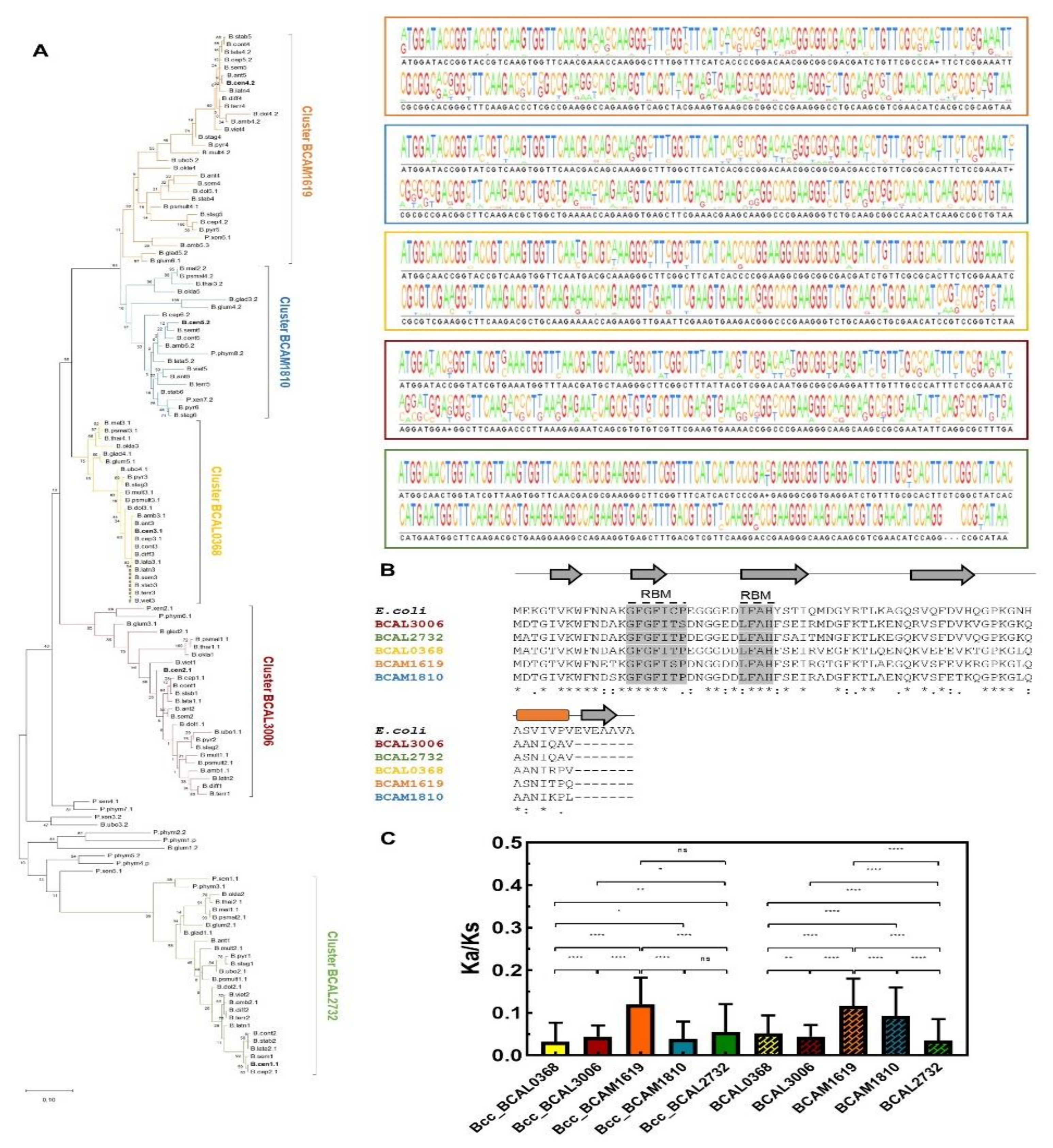
3.4. Cold Shock Proteins Encoded in Bcc Genomes
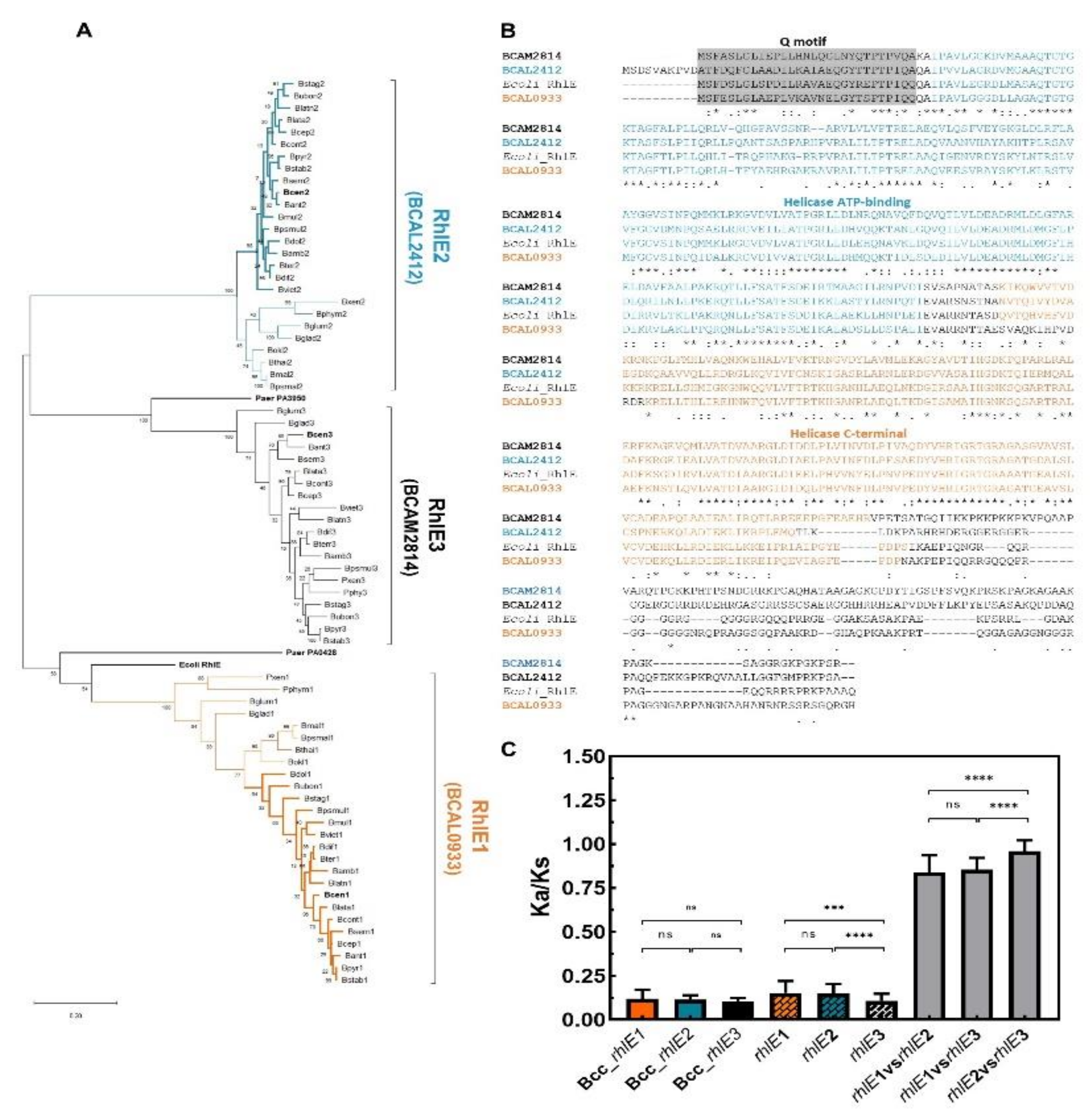
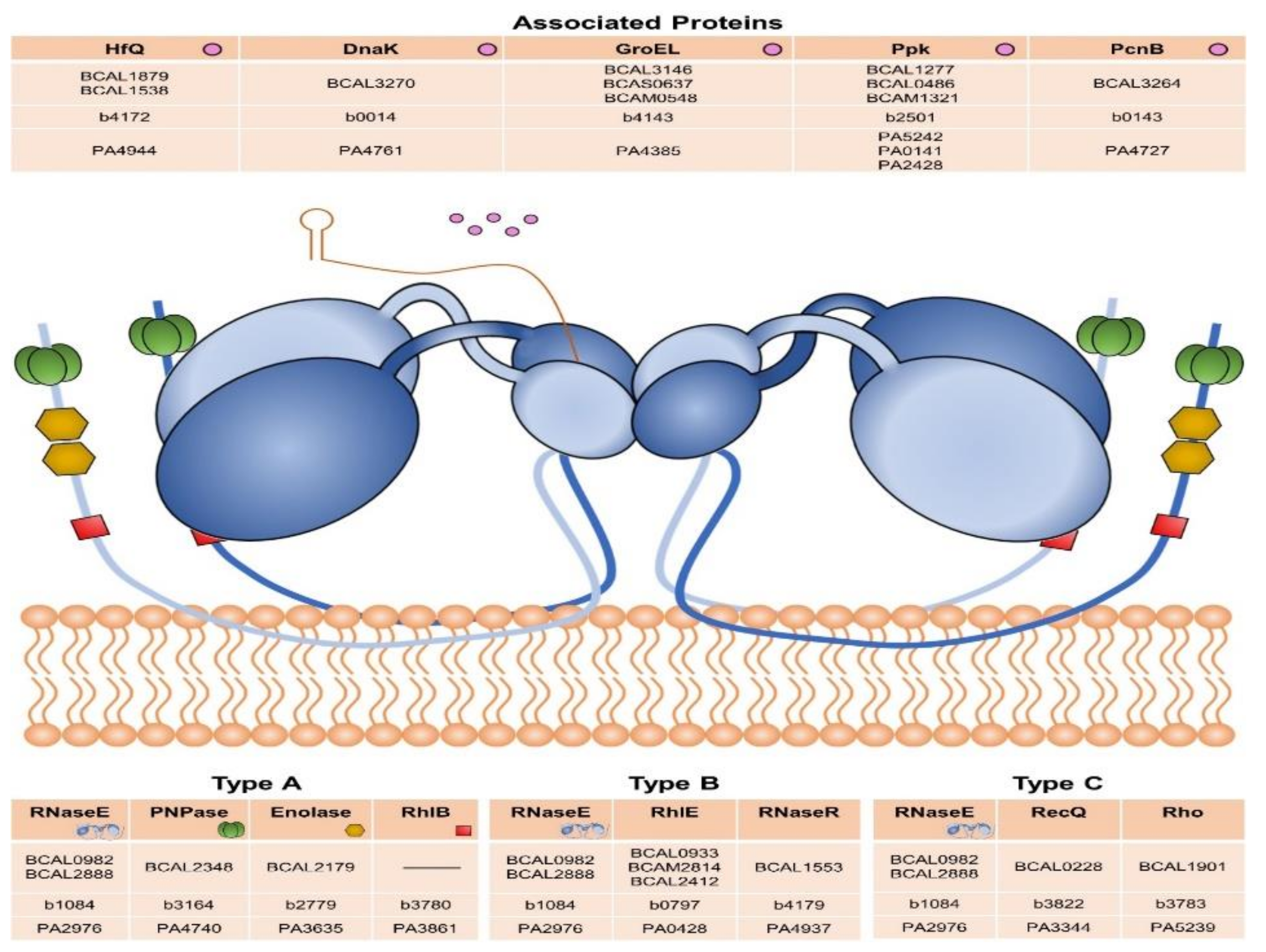
3.5. DEAD-Box RNA Helicases and Proteins Involved in RNA Degradation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gerstberger, S.; Hafner, M.; Tuschl, T. A Census of Human RNA-Binding Proteins. Nat. Rev. Genet. 2014, 15, 829. [Google Scholar] [CrossRef]
- Hentze, M.W.; Castello, A.; Schwarzl, T.; Preiss, T. A Brave New World of RNA-Binding Proteins. Nat. Rev. Mol. Cell Biol. 2018, 19, 327. [Google Scholar] [CrossRef]
- Helder, S.; Blythe, A.J.; Bond, C.S.; Mackay, J.P. Determinants of Affinity and Specificity in RNA-Binding Proteins. Curr. Opin. Struct. Biol. 2016, 38, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Holmqvist, E.; Vogel, J. RNA-Binding Proteins in Bacteria. Nat. Rev. Microbiol. 2018, 16, 601–615. [Google Scholar] [CrossRef] [PubMed]
- Van Assche, E.; Van Puyvelde, S.; Vanderleyden, J.; Steenackers, H.P. RNA-Binding Proteins Involved in Post-Transcriptional Regulation in Bacteria. Front. Microbiol. 2015, 6, 141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coppin, L.; Leclerc, J.; Vincent, A.; Porchet, N.; Pigny, P. Messenger RNA Life-Cycle in Cancer Cells: Emerging Role of Conventional and Non-Conventional RNA-Binding Proteins? Int. J. Mol. Sci. 2018, 19, 650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castello, A.; Fischer, B.; Frese, C.K.; Horos, R.; Alleaume, A.-M.; Foehr, S.; Curk, T.; Krijgsveld, J.; Hentze, M.W. Comprehensive Identification of RNA-Binding Domains in Human Cells. Mol. Cell 2016, 63, 696–710. [Google Scholar] [CrossRef] [Green Version]
- Smirnov, A.; Förstner, K.U.; Holmqvist, E.; Otto, A.; Günster, R.; Becher, D.; Reinhardt, R.; Vogel, J. Grad-Seq Guides the Discovery of ProQ as a Major Small RNA-Binding Protein. Proc. Natl. Acad. Sci. USA 2016, 113, 11591–11596. [Google Scholar] [CrossRef] [Green Version]
- Babitzke, P.; Lai, Y.-J.; Renda, A.J.; Romeo, T. Posttranscription Initiation Control of Gene Expression Mediated by Bacterial RNA-Binding Proteins. Annu. Rev. Microbiol. 2019, 73, 43–67. [Google Scholar] [CrossRef]
- Waters, L.S.; Storz, G. Regulatory RNAs in Bacteria. Cell 2009, 136, 615–628. [Google Scholar] [CrossRef] [Green Version]
- Gottesman, S.; Storz, G. Bacterial Small RNA Regulators: Versatile Roles and Rapidly Evolving Variations. Cold Spring Harb. Perspect. Biol. 2011, 3, a003798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hör, J.; Gorski, S.A.; Vogel, J. Bacterial RNA Biology on a Genome Scale. Mol. Cell 2018, 70, 785–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pita, T.; Feliciano, J.; Leitão, J. Small Noncoding Regulatory RNAs from Pseudomonas aeruginosa and Burkholderia cepacia complex. Int. J. Mol. Sci. 2018, 19, 3759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feliciano, J.R.; Grilo, A.M.; Guerreiro, S.I.; Sousa, S.A.; Leitão, J.H. Hfq: A Multifaceted RNA Chaperone Involved in Virulence. Future Microbiol. 2016, 11, 137–151. [Google Scholar] [CrossRef] [PubMed]
- Kusmierek, M.; Dersch, P. Regulation of Host–Pathogen Interactions via the Post-Transcriptional Csr/Rsm System. Curr. Opin. Microbiol. 2018, 41, 58–67. [Google Scholar] [CrossRef]
- Holmqvist, E.; Li, L.; Bischler, T.; Barquist, L.; Vogel, J. Global Maps of ProQ Binding in vivo Reveal Target Recognition via RNA Structure and Stability Control at mRNA 3′ Ends. Mol. Cell 2018, 70, 971–982. [Google Scholar] [CrossRef] [Green Version]
- Holmqvist, E.; Wright, P.R.; Li, L.; Bischler, T.; Barquist, L.; Reinhardt, R.; Backofen, R.; Vogel, J. Global RNA Recognition Patterns of Post-transcriptional Regulators Hfq and CsrA Revealed by UV Crosslinking in vivo. EMBO J. 2016, 35, 991–1011. [Google Scholar] [CrossRef]
- Sharan, M.; Förstner, K.U.; Eulalio, A.; Vogel, J. APRICOT: An Integrated Computational Pipeline for the Sequence-Based Identification and Characterization of RNA-Binding Proteins. Nucleic Acids Res. 2017, 45, e96. [Google Scholar] [CrossRef]
- Bressin, A.; Schulte-Sasse, R.; Figini, D.; Urdaneta, E.C.; Beckmann, B.M.; Marsico, A. TriPepSVM: De Novo Prediction of RNA-Binding Proteins Based on Short Amino Acid Motifs. Nucleic Acids Res. 2019, 47, 4406–4417. [Google Scholar] [CrossRef] [Green Version]
- Sousa, S.A.; Ramos, C.G.; Leitão, J.H. Burkholderia cepacia complex: Emerging Multihost Pathogens Equipped with a Wide Range of Virulence Factors and Determinants. Int. J. Microbiol. 2011, 2011. [Google Scholar] [CrossRef] [Green Version]
- Martina, P.; Leguizamon, M.; Prieto, C.I.; Sousa, S.A.; Montanaro, P.; Draghi, W.O.; Stämmler, M.; Bettiol, M.; de Carvalho, C.C.C.R.; Palau, J.; et al. Burkholderia puraquae sp. nov., a Novel Species of the Burkholderia cepacia complex Isolated from Hospital Settings and Agricultural Soils. Int. J. Syst. Evol. Microbiol. 2017, 68, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Vandamme, P.; Dawyndt, P. Classification and Identification of the Burkholderia cepacia complex: Past, Present and Future. Syst. Appl. Microbiol. 2011, 34, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Mahenthiralingam, E.; Urban, T.A.; Goldberg, J.B. The Multifarious, Multireplicon Burkholderia cepacia complex. Nat. Rev. Microbiol. 2005, 3, 144–156. [Google Scholar] [CrossRef] [PubMed]
- Yap, D.Y.H.; Choy, C.B.Y.; Mok, M.M.Y.; Wong, T.K.; Chan, T.M. Burkholderia cepacia-an Uncommon Cause of Exit-Site Infection in a Peritoneal Dialysis Patient. Perit. Dial. Int. 2014, 34, 471–472. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.Y.; Yong, D.; Lee, K.; Kim, H.S.; Kim, D.S. Burkholderia Sepsis in Children as a Hospital-Acquired Infection. Yonsei Med. J. 2016, 57, 97–102. [Google Scholar] [CrossRef]
- Sapkota, S.; Ganesan, S.K.; Bhattacharjee, S.; Mona, P.; Priyank, T.; Savitha, S.; Narsimhamurthy, R.; Naik, R. Clinical Spectrum of Burkholderia cepacia Infection in Cancer Patients: A Retrospective Study of an Emerging Disease. Med. Microbiol. Rep. 2019, 2, 1–4. [Google Scholar]
- Magalhães, M.; Doherty, C.; Govan, J.R.W.; Vandamme, P. Polyclonal Outbreak of Burkholderia cepacia complex Bacteraemia in Haemodialysis Patients. J. Hosp. Infect. 2003, 54, 120–123. [Google Scholar] [CrossRef]
- Sousa, S.A.; Feliciano, J.R.; Pita, T.; Guerreiro, S.I.; Leitão, J.H. Burkholderia cepacia complex Regulation of Virulence Gene Expression: A Review. Genes 2017, 8, 43. [Google Scholar] [CrossRef] [Green Version]
- Sousa, S.A.; Ramos, C.G.; Moreira, L.M.; Leitão, J.H. The Hfq Gene Is Required for Stress Resistance and Full Virulence of Burkholderia cepacia to the Nematode Caenorhabditis elegans. Microbiology 2010, 156 Pt 3, 896–908. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J.; et al. Pfam: The Protein Families Database. Nucleic Acids Res. 2014, 42, D222–D230. [Google Scholar] [CrossRef] [Green Version]
- Hunter, S.; Apweiler, R.; Attwood, T.K.; Bairoch, A.; Bateman, A.; Binns, D.; Bork, P.; Das, U.; Daugherty, L.; Duquenne, L.; et al. InterPro: The Integrative Protein Signature Database. Nucleic Acids Res. 2009, 37, D211–D215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binns, D.; Dimmer, E.; Huntley, R.; Barrell, D.; O’Donovan, C.; Apweiler, R. QuickGO: A Web-Based Tool for Gene Ontology Searching. Bioinformatics 2009, 25, 3045–3046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whiteside, M.D.; Winsor, G.L.; Laird, M.R.; Brinkman, F.S.L. OrtholugeDB: A Bacterial and Archaeal Orthology Resource for Improved Comparative Genomic Analysis. Nucleic Acids Res. 2012, 41, D366–D376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huerta-Cepas, J.; Szklarczyk, D.; Forslund, K.; Cook, H.; Heller, D.; Walter, M.C.; Rattei, T.; Mende, D.R.; Sunagawa, S.; Kuhn, M.; et al. EggNOG 4.5: A Hierarchical Orthology Framework with Improved Functional Annotations for Eukaryotic, Prokaryotic and Viral Sequences. Nucleic Acids Res. 2016, 44, D286–D293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Kriventseva, E.V.; Kuznetsov, D.; Tegenfeldt, F.; Manni, M.; Dias, R.; Simão, F.A.; Zdobnov, E.M. OrthoDB V10: Sampling the Diversity of Animal, Plant, Fungal, Protist, Bacterial and Viral Genomes for Evolutionary and Functional Annotations of Orthologs. Nucleic Acids Res. 2019, 47, D807–D811. [Google Scholar] [CrossRef] [Green Version]
- Winsor, G.L.; Khaira, B.; Van Rossum, T.; Lo, R.; Whiteside, M.D.; Brinkman, F.S.L. The Burkholderia Genome Database: Facilitating Flexible Queries and Comparative Analyses. Bioinformatics 2008, 24, 2803–2804. [Google Scholar] [CrossRef] [Green Version]
- Benson, D.A.; Cavanaugh, M.; Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2013, 41, D36–D42. [Google Scholar] [CrossRef] [Green Version]
- Consortium, T.U. Update on Activities at the Universal Protein Resource (UniProt) in 2013. Nucleic Acids Res. 2013, 41, D43–D47. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a Reference Resource for Gene and Protein Annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. MUSCLE: A Multiple Sequence Alignment Method with Reduced Time and Space Complexity. BMC Bioinform. 2004, 5, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.A.; Clamp, M.; Barton, G.J. Jalview Version 2-a Multiple Sequence Alignment Editor and Analysis Workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGuffin, L.J.; Bryson, K.; Jones, D.T. The PSIPRED Protein Structure Prediction Server. Bioinformatics 2000, 16, 404–405. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree of Life (ITOL) v3: An Online Tool for the Display and Annotation of Phylogenetic and Other Trees. Nucleic Acids Res. 2016, 44, W242–W245. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, J.; Zhao, X.-Q.; Wang, J.; Wong, G.K.-S.; Yu, J. KaKs_Calculator: Calculating Ka and Ks Through Model Selection and Model Averaging. Genom. Proteom. Bioinform. 2006, 4, 259–263. [Google Scholar] [CrossRef] [Green Version]
- Koonin, E.V.; Wolf, Y.I. Genomics of Bacteria and Archaea: The Emerging Dynamic View of the Prokaryotic World. Nucleic Acids Res. 2008, 36, 6688–6719. [Google Scholar] [CrossRef] [Green Version]
- Pandey, D.P.; Gerdes, K. Toxin-Antitoxin Loci Are Highly Abundant in Free-Living but Lost from Host-Associated Prokaryotes. Nucleic Acids Res. 2005, 33, 966–976. [Google Scholar] [CrossRef]
- Van Acker, H.; Sass, A.; Dhondt, I.; Nelis, H.J.; Coenye, T. Involvement of Toxin–Antitoxin Modules in Burkholderia cenocepacia Biofilm Persistence. Pathog. Dis. 2014, 71, 326–335. [Google Scholar] [CrossRef]
- Bloodworth, R.A.M.; Gislason, A.S.; Cardona, S.T. Burkholderia cenocepacia Conditional Growth Mutant Library Created by Random Promoter Replacement of Essential Genes. Microbiologyopen 2013, 2, 243–258. [Google Scholar] [CrossRef]
- Holden, M.T.G.; Seth-Smith, H.M.B.; Crossman, L.C.; Sebaihia, M.; Bentley, S.D.; Cerdeño-Tárraga, A.M.; Thomson, N.R.; Bason, N.; Quail, M.A.; Sharp, S.; et al. The Genome of Burkholderia cenocepacia J2315, an Epidemic Pathogen of Cystic Fibrosis Patients. J. Bacteriol. 2009, 191, 261–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sousa, S.A.; Ramos, C.G.; Almeida, F.; Meirinhos-Soares, L.; Wopperer, J.; Schwager, S.; Eberl, L.; Leitão, J.H. Burkholderia cenocepacia J2315 Acyl Carrier Protein: A Potential Target for Antimicrobials’ Development? Microb. Pathog. 2008, 45, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Melnikov, S.; Ben-Shem, A.; Garreau de Loubresse, N.; Jenner, L.; Yusupova, G.; Yusupov, M. One Core, Two Shells: Bacterial and Eukaryotic Ribosomes. Nat. Struct. Mol. Biol. 2012, 19, 560–567. [Google Scholar] [CrossRef] [PubMed]
- Yutin, N.; Puigbò, P.; Koonin, E.V.; Wolf, Y.I. Phylogenomics of Prokaryotic Ribosomal Proteins. PLoS ONE 2012, 7, e36972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sass, A.M.; Schmerk, C.; Agnoli, K.; Norville, P.J.; Eberl, L.; Valvano, M.A.; Mahenthiralingam, E. The Unexpected Discovery of a Novel Low-Oxygen-Activated Locus for the Anoxic Persistence of Burkholderia cenocepacia. ISME J. 2013, 14, 1568–1581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Duin, J.; Wijnands, R. The Function of Ribosomal Protein S21 in Protein Synthesis. Eur. J. Biochem. 1981, 118, 615–619. [Google Scholar] [CrossRef]
- Kimura, S.; Suzuki, T. Fine-Tuning of the Ribosomal Decoding Center by Conserved Methyl-Modifications in the Escherichia coli 16S rRNA. Nucleic Acids Res. 2010, 38, 1341–1352. [Google Scholar] [CrossRef] [Green Version]
- Deniziak, M.A.; Barciszewski, J. Methionyl-tRNA Synthetase. Acta Biochim. Pol. 2001, 48, 337–350. [Google Scholar] [CrossRef]
- Yi, H.; Lee, H.; Cho, K.-H.; Kim, H.S. Mutations in MetG (Methionyl-tRNA Synthetase) and TrmD [tRNA (Guanine-N1)-Methyltransferase] Conferring Meropenem Tolerance in Burkholderia thailandensis. J. Antimicrob. Chemother. 2017, 73, 332–338. [Google Scholar] [CrossRef] [Green Version]
- Sawana, A.; Adeolu, M.; Gupta, R.S. Molecular signatures and phylogenomic analysis of the genus Burkholderia: Proposal for division of this genus into the emended genus Burkholderia containing pathogenic organisms and a new genus Paraburkholderia gen. nov. harboring environmental species. Front. Genet. 2014, 5, 429. [Google Scholar] [CrossRef] [Green Version]
- Calabretta, S.; Richard, S. Emerging Roles of Disordered Sequences in RNA-Binding Proteins. Trends Biochem. Sci. 2015, 40, 662–672. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Cepas, J.; Capella-Gutierrez, S.; Pryszcz, L.P.; Denisov, I.; Kormes, D.; Marcet-Houben, M.; Gabaldón, T. PhylomeDB v3.0: An Expanding Repository of Genome-Wide Collections of Trees, Alignments and Phylogeny-Based Orthology and Paralogy Predictions. Nucleic Acids Res. 2011, 39 (Suppl. 1), D556–D560. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Zhulin, I.; Wartell, R.M. Predicted Structure and Phyletic Distribution of the RNA-Binding Protein Hfq. Nucleic Acids Res. 2002, 30, 3662–3671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, T.; Keto-Timonen, R.; Jiang, X.; Virtanen, J.-P.; Korkeala, H. Insights into the Phylogeny and Evolution of Cold Shock Proteins: From Enteropathogenic Yersinia and Escherichia coli to Eubacteria. Int. J. Mol. Sci. 2019, 20, 4059. [Google Scholar] [CrossRef] [Green Version]
- Horn, G.; Hofweber, R.; Kremer, W.; Kalbitzer, H.R. Structure and Function of Bacterial Cold Shock Proteins. Cell. Mol. Life Sci. 2007, 64, 1457. [Google Scholar] [CrossRef]
- Yamanaka, K.; Inouye, M. Growth-Phase-Dependent Expression of CspD, Encoding a Member of the CspA Family in Escherichia coli. J. Bacteriol. 1997, 179, 5126–5130. [Google Scholar] [CrossRef] [Green Version]
- Yamanaka, K.; Zheng, W.; Crooke, E.; Wang, Y.-H.; Inouye, M. CspD, a Novel DNA Replication Inhibitor Induced during the Stationary Phase in Escherichia coli. Mol. Microbiol. 2001, 39, 1572–1584. [Google Scholar] [CrossRef]
- Kim, Y.; Wood, T.K. Toxins Hha and CspD and Small RNA Regulator Hfq Are Involved in Persister Cell Formation through MqsR in Escherichia coli. Biochem. Biophys. Res. Commun. 2010, 391, 209–213. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Wang, X.; Zhang, X.-S.; Grigoriu, S.; Page, R.; Peti, W.; Wood, T.K. Escherichia coli Toxin/Antitoxin Pair MqsR/MqsA Regulate Toxin CspD. Environ. Microbiol. 2010, 12, 1105–1121. [Google Scholar] [CrossRef] [Green Version]
- Cordin, O.; Banroques, J.; Tanner, N.K.; Linder, P. The DEAD-Box Protein Family of RNA Helicases. Gene 2006, 367, 17–37. [Google Scholar] [CrossRef]
- Prud’homme-Généreux, A.; Beran, R.K.; Iost, I.; Ramey, C.S.; Mackie, G.A.; Simons, R.W. Physical and Functional Interactions among RNase E, Polynucleotide Phosphorylase and the Cold-Shock Protein, CsdA: Evidence for a ‘Cold Shock Degradosome’. Mol. Microbiol. 2004, 54, 1409–1421. [Google Scholar] [CrossRef] [PubMed]
- Raynal, L.C.; Carpousis, A.J. Poly(A) Polymerase I of Escherichia coli: Characterization of the Catalytic Domain, an RNA Binding Site and Regions for the Interaction with Proteins Involved in mRNA Degradation. Mol. Microbiol. 1999, 32, 765–775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charollais, J.; Dreyfus, M.; Iost, I. CsdA, a Cold-shock RNA Helicase from Escherichia coli, Is Involved in the Biogenesis of 50S Ribosomal Subunit. Nucleic Acids Res. 2004, 32, 2751–2759. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Aoki, H.; Ganoza, M.C. Molecular Characterization of a Prokaryotic Translation Factor Homologous to the Eukaryotic Initiation Factor EIF4A. Int. J. Biochem. Cell Biol. 1999, 31, 215–229. [Google Scholar] [CrossRef]
- Sharpe Elles, L.M.; Sykes, M.T.; Williamson, J.R.; Uhlenbeck, O.C. A Dominant Negative Mutant of the E. coli RNA Helicase DbpA Blocks Assembly of the 50S Ribosomal Subunit. Nucleic Acids Res. 2009, 37, 6503–6514. [Google Scholar] [CrossRef] [Green Version]
- Tsu, C.A.; Kossen, K.; Uhlenbeck, O.C. The Escherichia coli DEAD Protein DbpA Recognizes a Small RNA Hairpin in 23S rRNA. RNA 2001, 7, 702–709. [Google Scholar] [CrossRef] [Green Version]
- Diges, C.M.; Uhlenbeck, O.C. Escherichia coli DbpA Is a 3‘ → 5‘ RNA Helicase. Biochemistry 2005, 44, 7903–7911. [Google Scholar] [CrossRef]
- Py, B.; Higgins, C.F.; Krisch, H.M.; Carpousis, A.J. A DEAD-Box RNA Helicase in the Escherichia coli RNA Degradosome. Nature 1996, 381, 169–172. [Google Scholar] [CrossRef]
- Khemici, V.; Poljak, L.; Toesca, I.; Carpousis, A.J. Evidence in vivo That the DEAD-Box RNA Helicase RhlB Facilitates the Degradation of Ribosome-Free mRNA by RNase E. Proc. Natl. Acad. Sci. USA 2005, 102, 6913–6918. [Google Scholar] [CrossRef] [Green Version]
- Chandran, V.; Poljak, L.; Vanzo, N.F.; Leroy, A.; Miguel, R.N.; Fernandez-Recio, J.; Parkinson, J.; Burns, C.; Carpousis, A.J.; Luisi, B.F. Recognition and Cooperation Between the ATP-Dependent RNA Helicase RhlB and Ribonuclease RNase E. J. Mol. Biol. 2007, 367, 113–132. [Google Scholar] [CrossRef]
- Jain, C. The E. coli RhlE RNA Helicase Regulates the Function of Related RNA Helicases during Ribosome Assembly. RNA 2008, 14, 381–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iost, I.; Dreyfus, M. mRNAs Can Be Stabilized by DEAD-Box Proteins. Nature 1994, 372, 193–196. [Google Scholar] [CrossRef] [PubMed]
- Charollais, J.; Pflieger, D.; Vinh, J.; Dreyfus, M.; Iost, I. The DEAD-Box RNA Helicase SrmB Is Involved in the Assembly of 50S Ribosomal Subunits in Escherichia coli. Mol. Microbiol. 2003, 48, 1253–1265. [Google Scholar] [CrossRef]
- Janda, J.M.; Abbott, S.L. The Genus Shewanella: From the Briny Depths below to Human Pathogen. Crit. Rev. Microbiol. 2014, 40, 293–312. [Google Scholar] [CrossRef] [PubMed]
- Redder, P.; Hausmann, S.; Khemici, V.; Yasrebi, H.; Linder, P. Bacterial Versatility Requires DEAD-Box RNA Helicases. FEMS Microbiol. Rev. 2015, 39, 392–412. [Google Scholar] [CrossRef] [PubMed]
- Awano, N.; Xu, C.; Ke, H.; Inoue, K.; Inouye, M.; Phadtare, S. Complementation Analysis of the Cold-Sensitive Phenotype of the Escherichia coli CsdA Deletion Strain. J. Bacteriol. 2007, 189, 5808–5815. [Google Scholar] [CrossRef] [Green Version]
- Silverman, E.; Edwalds-Gilbert, G.; Lin, R.-J. DExD/H-Box Proteins and Their Partners: Helping RNA Helicases Unwind. Gene 2003, 312, 1–16. [Google Scholar] [CrossRef]
- Khemici, V.; Toesca, I.; Poljak, L.; Vanzo, N.F.; Carpousis, A.J. The RNase E of Escherichia coli has at Least Two Binding Sites for DEAD-Box RNA Helicases: Functional Replacement of RhlB by RhlE. Mol. Microbiol. 2004, 54, 1422–1430. [Google Scholar] [CrossRef]
- Purusharth, R.I.; Klein, F.; Sulthana, S.; Jäger, S.; Jagannadham, M.V.; Evguenieva-Hackenberg, E.; Ray, M.K.; Klug, G. Exoribonuclease R Interacts with Endoribonuclease E and an RNA Helicase in the Psychrotrophic Bacterium Pseudomonas syringae Lz4W. J. Biol. Chem. 2005, 280, 14572–14578. [Google Scholar] [CrossRef] [Green Version]
- Jäger, S.; Fuhrmann, O.; Heck, C.; Hebermehl, M.; Schiltz, E.; Rauhut, R.; Klug, G. An mRNA Degrading Complex in Rhodobacter capsulatus. Nucleic Acids Res. 2001, 29, 4581–4588. [Google Scholar] [CrossRef]
- Zheng, J.; Zhang, X.; Zhao, X.; Tong, X.; Hong, X.; Xie, J.; Liu, S. Deep-RBPPred: Predicting RNA binding proteins in the proteome scale based on deep learning. Sci. Rep. 2018, 8, 15264. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DEAD-Box RNA Helicases | mRNA Processing and Decay | Ribosome Biogenesis | Translation Initiation |
|---|---|---|---|
| DeaD | In vivo formation of a cold-shock degradosome with RNase E [71]; In vitro interaction with E. coli poly(A) polymerase [72]. | Involved in 50S ribosomal subunit assembly, acting after SrmB [73]; Putative role in the biogenesis of the 30S ribosomal subunit [73]. | Stimulates translation of some mRNAs, probably at the level of initiation [74]. |
| DbpA | Involved in the assembly of the 50S ribosomal subunit [75]; RNA-dependent ATPase activity specific for 23S rRNA [76]; 3′ to 5′ RNA helicase activity that uses the energy of ATP hydrolysis to destabilize and unwind short rRNA duplexes [77]. | ||
| RhlB | Component of the RNA degradosome. Interaction with RNase E and co-localization with it at the membrane [78]; Facilitates RNase E cleavage of ribosome-free mRNA and highly RNase E-sensitive mRNAs [79]; RhlB activity prefers a 5’ single-stranded extension in presence of a fragment of RNase E (aa 628–843) [80] | ||
| RhlE | Interaction with RNase E [79]; In vitro interaction with E. coli poly(A) polymerase [72]; Ability to unwind a short blunt-ended RNA duplex. | May play a role in the interconversion of ribosomal RNA-folding intermediates that are further processed by DeaD or SrmB during ribosome maturation [81]. | |
| SrmB | Stabilization of certain mRNAs [82]. Interaction with RNase E [79] In vitro interaction with E. coli poly(A) polymerase [72]. | Assembly of the 50S ribosomal subunit at low temperature, acting before DeaD [83]. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feliciano, J.R.; Seixas, A.M.M.; Pita, T.; Leitão, J.H. Comparative Genomics and Evolutionary Analysis of RNA-Binding Proteins of Burkholderia cenocepacia J2315 and Other Members of the B. cepacia Complex. Genes 2020, 11, 231. https://doi.org/10.3390/genes11020231
Feliciano JR, Seixas AMM, Pita T, Leitão JH. Comparative Genomics and Evolutionary Analysis of RNA-Binding Proteins of Burkholderia cenocepacia J2315 and Other Members of the B. cepacia Complex. Genes. 2020; 11(2):231. https://doi.org/10.3390/genes11020231
Chicago/Turabian StyleFeliciano, Joana R., António M. M. Seixas, Tiago Pita, and Jorge H. Leitão. 2020. "Comparative Genomics and Evolutionary Analysis of RNA-Binding Proteins of Burkholderia cenocepacia J2315 and Other Members of the B. cepacia Complex" Genes 11, no. 2: 231. https://doi.org/10.3390/genes11020231
APA StyleFeliciano, J. R., Seixas, A. M. M., Pita, T., & Leitão, J. H. (2020). Comparative Genomics and Evolutionary Analysis of RNA-Binding Proteins of Burkholderia cenocepacia J2315 and Other Members of the B. cepacia Complex. Genes, 11(2), 231. https://doi.org/10.3390/genes11020231