From Genes to -Omics: The Evolving Molecular Landscape of Malignant Peripheral Nerve Sheath Tumor


Abstract
:1. Clinical Overview of MPNST
2. Germline Loss of NF1: Correlations with NF1 Phenotype
3. Sequencing Efforts in Human MPNST Samples: Improvements in Technology with Variability in Study Design
4. Somatic NF1 Mutations in Tumors Including MPNST
5. Acquired Mutations during Transformation from pNF
5.1. Loss of CDKN2A/B: Correlations with the pNF to ANF Transition
5.2. LOH and Mutation in the Tumor Suppressor TP53: Not Universal in Human MPNST
5.3. Loss of PRC2 or H3K27me3: Recurrently and Specifically Occurs in MPNST
6. Less Common Recurrent Variants Identified with Modern Sequencing Investigations of MPNST
6.1. BRAF Mutation: An Alternate Mechanism for Activation of RAS Signaling
6.2. EGFR, MET and Other Receptor Tyrosine Kinases: Frequent Copy Number Gains in MPNST
6.3. AURKA Amplification
6.4. Tyrosine Kinase 2 Overexpression in MPNST
6.5. ATRX Mutation and Evidence for Alternative Lengthening of Telomeres
6.6. Beyond SUZ12: Less Common Variant Mutations in Other Chromatin Modifying Genes
6.7. Evidence for Alterations in the HIPPO Pathway in a Subset of MPNST and Schwann Cell Derived Tumors
7. Beyond Genomics: The State of Understanding MPNST Transcriptomes, Proteomes, Epigenomes, and Metabolomes
8. Translating Molecular Landscape of MPNST into Improved Therapies for Patients
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Carroll, S.L. Molecular mechanisms promoting the pathogenesis of Schwann cell neoplasms. Acta Neuropathol. 2012, 123, 321–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Ghosh, P.; Charnay, P.; Burns, D.K.; Parada, L.F. Neurofibromas in NF1: Schwann cell origin and role of tumor environment. Science 2002, 296, 920–922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferner, R.E.; Gutmann, D.H. International consensus statement on malignant peripheral nerve sheath tumors in neurofibromatosis. Cancer Res. 2002, 62, 1573–1577. [Google Scholar]
- Evans, D.G.; Baser, M.E.; McGaughran, J.; Sharif, S.; Howard, E.; Moran, A. Malignant peripheral nerve sheath tumours in neurofibromatosis 1. J. Med. Genet. 2002, 39, 311–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCaughan, J.A.; Holloway, S.M.; Davidson, R.; Lam, W.W. Further evidence of the increased risk for malignant peripheral nerve sheath tumour from a Scottish cohort of patients with neurofibromatosis type 1. J. Med. Genet. 2007, 44, 463–466. [Google Scholar] [CrossRef] [PubMed]
- Kattan, M.W.; Leung, D.H.; Brennan, M.F. Postoperative nomogram for 12-year sarcoma-specific death. J. Clin. Oncol. 2002, 20, 791–796. [Google Scholar] [CrossRef]
- Fletcher, C.D.; McKee, P.H. Sarcomas—A clinicopathological guide with particular reference to cutaneous manifestation. II. Malignant nerve sheath tumour, leiomyosarcoma and rhabdomyosarcoma. Clin. Exp. Dermatol 1985, 10, 201–216. [Google Scholar] [CrossRef]
- Higham, C.S.; Steinberg, S.M.; Dombi, E.; Perry, A.; Helman, L.J.; Schuetze, S.M.; Ludwig, J.A.; Staddon, A.; Milhem, M.M.; Rushing, D.; et al. SARC006: Phase II Trial of Chemotherapy in Sporadic and Neurofibromatosis Type 1 Associated Chemotherapy-Naive Malignant Peripheral Nerve Sheath Tumors. Sarcoma 2017, 2017, 8685638. [Google Scholar] [CrossRef] [Green Version]
- Gutmann, D.H.; Ferner, R.E.; Listernick, R.H.; Korf, B.R.; Wolters, P.L.; Johnson, K.J. Neurofibromatosis type 1. Nat. Rev. Dis. Primers 2017, 3, 17004. [Google Scholar] [CrossRef]
- Ly, K.I.; Blakeley, J.O. The Diagnosis and Management of Neurofibromatosis Type 1. Med. Clin. N. Am. 2019, 103, 1035–1054. [Google Scholar] [CrossRef]
- Uusitalo, E.; Rantanen, M.; Kallionpää, R.A.; Poyhonen, M.; Leppavirta, J.; Yla-Outinen, H.; Riccardi, V.M.; Pukkala, E.; Pitkaniemi, J.; Peltonen, S.; et al. Distinctive Cancer Associations in Patients with Neurofibromatosis Type 1. J. Clin. Oncol. 2016, 34, 1978–1986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gross, A.M.; Singh, G.; Akshintala, S.; Baldwin, A.; Dombi, E.; Ukwuani, S.; Goodwin, A.; Liewehr, D.J.; Steinberg, S.M.; Widemann, B.C. Association of Plexiform Neurofibroma Volume Changes and Development of Clinical Morbidities in Neurofibromatosis 1. Neuro. Oncol. 2018, 12, 1643–1651. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.; Stewart, D.R.; Reilly, K.M.; Viskochil, D.; Miettinen, M.M.; Widemann, B.C. Malignant Peripheral Nerve Sheath Tumors State of the Science: Leveraging Clinical and Biological Insights into Effective Therapies. Sarcoma 2017, 2017, 7429697. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.H.; Harper, P.S.; Upadhyaya, M. Molecular genetics of neurofibromatosis type 1 (NF1). J. Med. Genet. 1996, 33, 2–17. [Google Scholar] [CrossRef] [Green Version]
- Jett, K.; Friedman, J.M. Clinical and genetic aspects of neurofibromatosis 1. Genet. Med. 2010, 12, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Anderson, J.L.; Gutmann, D.H. Neurofibromatosis type 1. Handb. Clin. Neurol. 2015, 132, 75–86. [Google Scholar]
- Wallace, M.R.; Marchuk, D.A.; Andersen, L.B.; Letcher, R.; Odeh, H.M.; Saulino, A.M.; Fountain, J.W.; Brereton, A.; Nicholson, J.; Mitchell, A.L.; et al. Type 1 neurofibromatosis gene: Identification of a large transcript disrupted in three NF1 patients. Science 1990, 249, 181–186. [Google Scholar] [CrossRef]
- Andersen, L.B.; Ballester, R.; Marchuk, D.A.; Chang, E.; Gutmann, D.H.; Saulino, A.M.; Camonis, J.; Wigler, M.; Collins, F.S. A conserved alternative splice in the von Recklinghausen neurofibromatosis (NF1) gene produces two neurofibromin isoforms, both of which have GTPase-activating protein activity. Mol. Cell Biol. 1993, 13, 487–495. [Google Scholar] [CrossRef] [Green Version]
- Gutmann, D.H.; Wood, D.L.; Collins, F.S. Identification of the neurofibromatosis type 1 gene product. Proc. Natl. Acad. Sci. USA 1991, 88, 9658–9662. [Google Scholar] [CrossRef] [Green Version]
- DeClue, J.E.; Cohen, B.D.; Lowy, D.R. Identification and characterization of the neurofibromatosis type 1 protein product. Proc. Natl. Acad. Sci. USA 1991, 88, 9914–9918. [Google Scholar] [CrossRef] [Green Version]
- Martin, G.A.; Viskochil, D.; Bollag, G.; McCabe, P.C.; Crosier, W.J.; Haubruck, H.; Conroy, L.; Clark, R.; O′Connell, P.; Cawthon, R.M.; et al. The GAP-related domain of the neurofibromatosis type 1 gene product interacts with ras p21. Cell 1990, 63, 843–849. [Google Scholar] [CrossRef]
- Ballester, R.; Marchuk, D.; Boguski, M.; Letcher, R.; Wigler, M.; Collins, F. The NF1 locus encodes a protein functionally related to mammalian GAP and yeast IRA proteins. Cell 1990, 63, 851–859. [Google Scholar] [CrossRef] [Green Version]
- Basu, T.N.; Gutmann, D.H.; Fletcher, J.A.; Glover, T.W.; Collins, F.S.; Downward, J. Aberrant regulation of ras proteins in malignant tumour cells from type 1 neurofibromatosis patients. Nature 1992, 356, 713–715. [Google Scholar] [CrossRef] [PubMed]
- Bollag, G.; Clapp, D.W.; Shih, S.; Adler, F.; Zhang, Y.Y.; Thompson, P.; Lange, B.J.; Freedman, M.H.; McCormick, F.; Jacks, T.; et al. Loss of NF1 results in activation of the Ras signaling pathway and leads to aberrant growth in haematopoietic cells. Nat. Genet. 1996, 12, 144–148. [Google Scholar] [CrossRef] [PubMed]
- Cichowski, K.; Jacks, T. NF1 tumor suppressor gene function: Narrowing the GAP. Cell 2001, 104, 593–604. [Google Scholar] [CrossRef] [Green Version]
- Rosser, T.; Packer, R.J. Neurofibromas in children with neurofibromatosis 1. J. Child Neurol. 2002, 17, 585–651. [Google Scholar] [CrossRef]
- Messiaen, L.M.; Callens, T.; Mortier, G.; Beysen, D.; Vandenbroucke, I.; Van Roy, N.; Speleman, F.; Paepe, A.D. Exhaustive mutation analysis of the NF1 gene allows identification of 95% of mutations and reveals a high frequency of unusual splicing defects. Hum. Mutat. 2000, 15, 541–555. [Google Scholar] [CrossRef]
- Koczkowska, M.; Callens, T.; Gomes, A.; Sharp, A.; Chen, Y.; Hicks, A.D.; Aylsworth, A.S.; Azizi, A.A.; Basel, D.G.; Bellus, G.; et al. Expanding the clinical phenotype of individuals with a 3-bp in-frame deletion of the NF1 gene (c.2970_2972del): An update of genotype-phenotype correlation. Genet. Med. 2019, 21, 867–876. [Google Scholar] [CrossRef] [Green Version]
- Upadhyaya, M.; Huson, S.M.; Davies, M.; Thomas, N.; Chuzhanova, N.; Giovannini, S.; Evans, D.G.; Howard, E.; Kerr, B.; Griffiths, S.; et al. An absence of cutaneous neurofibromas associated with a 3-bp inframe deletion in exon 17 of the NF1 gene (c.2970-2972 delAAT): Evidence of a clinically significant NF1 genotype-phenotype correlation. Am. J. Hum. Genet. 2007, 80, 140–151. [Google Scholar] [CrossRef] [Green Version]
- Rojnueangnit, K.; Xie, J.; Gomes, A.; Sharp, A.; Callens, T.; Chen, Y.; Liu, Y.; Cochran, M.; Abbott, M.A.; Atkin, J.; et al. High Incidence of Noonan Syndrome Features Including Short Stature and Pulmonic Stenosis in Patients carrying NF1 Missense Mutations Affecting p.Arg1809: Genotype-Phenotype Correlation. Hum. Mutat. 2015, 36, 1052–1063. [Google Scholar] [CrossRef] [Green Version]
- De Raedt, T.; Maertens, O.; Chmaram, M.; Brems, H.; Heyns, I.; Sciot, R.; Majounie, E.; Upadhyaya, M.; De Schepper, S.; Speleman, F.; et al. Somatic loss of wild type NF1 allele in neurofibromas: Comparison of NF1 microdeletion and non-microdeletion patients. Genes Chromosomes Cancer 2006, 45, 893–904. [Google Scholar] [CrossRef] [PubMed]
- Bottillo, I.; Ahlquist, T.; Brekke, H.; Danielsen, S.A.; van den Berg, E.; Mertens, F.; Lothe, R.A.; Dallapiccola, B. Germline and somatic NF1 mutations in sporadic and NF1-associated malignant peripheral nerve sheath tumours. J. Pathol. 2009, 217, 693–701. [Google Scholar] [CrossRef] [PubMed]
- Pasmant, E.; Sabbagh, A.; Spurlock, G.; Laurendeau, I.; Grillo, E.; Hamel, M.J.; Martin, L.; Barbarot, S.; Leheup, B.; Rodriguez, D.; et al. NF1 microdeletions in neurofibromatosis type 1: From genotype to phenotype. Hum. Mutat. 2010, 31, E1506–E1518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kehrer-Sawatzki, H.; Mautner, V.F.; Cooper, D.N. Emerging genotype-phenotype relationships in patients with large NF1 deletions. Hum. Genet. 2017, 136, 349–376. [Google Scholar] [CrossRef] [Green Version]
- De Raedt, T.; Brems, H.; Wolkenstein, P.; Vidaud, D.; Pilotti, S.; Perrone, F.; Mautner, V.; Frahm, S.; Sciot, R.; Legius, E. Elevated risk for MPNST in NF1 microdeletion patients. Am. J. Hum. Genet. 2003, 72, 1288–1292. [Google Scholar] [CrossRef] [Green Version]
- Koczkowska, M.; Chen, Y.; Callens, T.; Gomes, A.; Sharp, A.; Johnson, S.; Hsiao, M.C.; Chen, Z.; Balasubramanian, M.; Barnett, C.P.; et al. Genotype-Phenotype Correlation in NF1: Evidence for a More Severe Phenotype Associated with Missense Mutations Affecting NF1 Codons 844–848. Am. J. Hum. Genet. 2018, 102, 69–87. [Google Scholar] [CrossRef]
- Bridge, R.S., Jr.; Bridge, J.A.; Neff, J.R.; Naumann, S.; Althof, P.; Bruch, L.A. Recurrent chromosomal imbalances and structurally abnormal breakpoints within complex karyotypes of malignant peripheral nerve sheath tumour and malignant triton tumour: A cytogenetic and molecular cytogenetic study. J. Clin. Pathol. 2004, 57, 1172–1178. [Google Scholar] [CrossRef]
- Mantripragada, K.K.; Spurlock, G.; Kluwe, L.; Chuzhanova, N.; Ferner, R.E.; Frayling, I.M.; Dumanski, J.P.; Guha, A.; Mautner, V.; Upadhyaya, M. High-resolution DNA copy number profiling of malignant peripheral nerve sheath tumors using targeted microarray-based comparative genomic hybridization. Clin. Cancer Res. 2008, 14, 1015–1024. [Google Scholar] [CrossRef] [Green Version]
- Verdijk, R.M.; den Bakker, M.A.; Dubbink, H.J.; Hop, W.C.; Dinjens, W.N.; Kros, J.M. TP53 mutation analysis of malignant peripheral nerve sheath tumors. J. Neuropathol. Exp. Neurol. 2010, 69, 16–26. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Ylipää, A.; Sun, Y.; Zheng, H.; Chen, K.; Nykter, M.; Trent, J.; Ratner, N.; Lev, D.C.; Zhang, W. Genomic and molecular characterization of malignant peripheral nerve sheath tumor identifies the IGF1R pathway as a primary target for treatment. Clin. Cancer Res. 2011, 17, 7563–7573. [Google Scholar] [CrossRef] [Green Version]
- De Raedt, T.; Beert, E.; Pasmant, E.; Luscan, A.; Brems, H.; Ortonne, N.; Helin, K.; Hornick, J.L.; Mautner, V.; Kehrer-Sawatzki, H.; et al. PRC2 loss amplifies Ras-driven transcription and confers sensitivity to BRD4-based therapies. Nature 2014, 514, 247–251. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Wang, Y.; Jones, S.; Sausen, M.; McMahon, K.; Sharma, R.; Wang, Q.; Belzberg, A.J.; Chaichana, K.; Gallia, G.L.; et al. Somatic mutations of SUZ12 in malignant peripheral nerve sheath tumors. Nat. Genet. 2014, 46, 1170–1172. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.; Teckie, S.; Wiesner, T.; Prieto Granada, C.N.; Lin, M.; Zhu, S.; Cao, Z.; Liang, Y.; Sboner, A.; Tap, W.D.; et al. PRC2 is recurrently inactivated through EED or SUZ12 loss in malignant peripheral nerve sheath tumors. Nat. Genet. 2014, 46, 1227–1232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sohier, P.; Luscan, A.; Lloyd, A.; Ashelford, K.; Laurendeau, I.; Briand-Suleau, A.; Vidaud, D.; Ortonne, N.; Pasmant, E.; Upadhyaya, M. Confirmation of mutation landscape of NF1-associated malignant peripheral nerve sheath tumors. Genes Chromosomes Cancer 2017, 56, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Brohl, A.S.; Kahen, E.; Yoder, S.J.; Teer, J.K.; Reed, D.R. The genomic landscape of malignant peripheral nerve sheath tumors: Diverse drivers of Ras pathway activation. Sci. Rep. 2017, 7, 14992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zehir, A.; Benayed, R.; Shah, R.H.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M.; et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 2017, 23, 703–713. [Google Scholar] [CrossRef]
- Kaplan, H.G.; Rostad, S.; Ross, J.S.; Ali, S.M.; Millis, S.Z. Genomic Profiling in Patients with Malignant Peripheral Nerve Sheath Tumors Reveals Multiple Pathways with Targetable Mutations. J. Natl. Compr. Cancer Netw. 2018, 16, 967–974. [Google Scholar] [CrossRef] [Green Version]
- Pemov, A.; Hansen, N.F.; Sindiri, S.; Patidar, R.; Higham, C.S.; Dombi, E.; Miettinen, M.M.; Fetsch, P.; Brems, H.; Chandrasekharappa, S.; et al. Low mutation burden and frequent loss of CDKN2A/B and SMARCA2, but not PRC2, define pre-malignant neurofibromatosis type 1-associated atypical neurofibromas. Neuro. Oncol. 2019, 21, 981–992. [Google Scholar] [CrossRef]
- Pollard, K.; Banerjee, J.; Doan, X.; Wang, J.; Guo, X.; Allaway, R.; Langmead, S.; Slobogean, B.; Meyer, C.F.; Loeb, D.M.; et al. A clinically and genomically annotated nerve sheath tumor biospecimen repository. Sci. Data 2020, 7, 184. [Google Scholar] [CrossRef]
- Xu, W.; Mulligan, L.M.; Ponder, M.A.; Liu, L.; Smith, B.A.; Mathew, C.G.; Ponder, B.A. Loss of NF1 alleles in phaeochromocytomas from patients with type I neurofibromatosis. Genes Chromosomes Cancer 1992, 4, 337–342. [Google Scholar] [CrossRef]
- McPherson, J.R.; Ong, C.K.; Ng, C.C.; Rajasegaran, V.; Heng, H.L.; Yu, W.S.; Tan, B.K.; Madhukumar, P.; Teo, M.C.; Ngeow, J.; et al. Whole-exome sequencing of breast cancer, malignant peripheral nerve sheath tumor and neurofibroma from a patient with neurofibromatosis type 1. Cancer Med. 2015, 4, 1871–1878. [Google Scholar] [CrossRef] [PubMed]
- Shannon, K.M.; O′Connell, P.; Martin, G.A.; Paderanga, D.; Olson, K.; Dinndorf, P.; McCormick, F. Loss of the normal NF1 allele from the bone marrow of children with type 1 neurofibromatosis and malignant myeloid disorders. N. Engl. J. Med. 1994, 330, 597–601. [Google Scholar] [CrossRef] [PubMed]
- Colman, S.D.; Williams, C.A.; Wallace, M.R. Benign neurofibromas in type 1 neurofibromatosis (NF1) show somatic deletions of the NF1 gene. Nat. Genet. 1995, 11, 90–92. [Google Scholar] [CrossRef]
- Serra, E.; Puig, S.; Otero, D.; Gaona, A.; Kruyer, H.; Ars, E.; Estivill, X.; Lázaro, C. Confirmation of a double-hit model for the NF1 gene in benign neurofibromas. Am. J. Hum. Genet. 1997, 61, 512–519. [Google Scholar] [CrossRef] [Green Version]
- Upadhyaya, M.; Kluwe, L.; Spurlock, G.; Monem, B.; Majounie, E.; Mantripragada, K.; Ruggieri, M.; Chuzhanova, N.; Evans, D.G.; Ferner, R.; et al. Germline and somatic NF1 gene mutation spectrum in NF1-associated malignant peripheral nerve sheath tumors (MPNSTs). Hum. Mutat. 2008, 29, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.C.; Ingram, D.A.; Chen, S.; Zhu, Y.; Yuan, J.; Li, X.; Yang, X.; Knowles, S.; Horn, W.; Li, Y.; et al. Nf1-dependent tumors require a microenvironment containing Nf1+/− and c-kit-dependent bone marrow. Cell 2008, 135, 437–448. [Google Scholar] [CrossRef] [Green Version]
- Beert, E.; Brems, H.; Daniëls, B.; De Wever, I.; Van Calenbergh, F.; Schoenaers, J.; Debiec-Rychter, M.; Gevaert, O.; De Raedt, T.; Van Den Bruel, A.; et al. Atypical neurofibromas in neurofibromatosis type 1 are premalignant tumors. Genes Chromosomes Cancer 2011, 50, 1021–1032. [Google Scholar] [CrossRef]
- Higham, C.S.; Dombi, E.; Rogiers, A.; Bhaumik, S.; Pans, S.; Connor, S.; Miettinen, M.; Sciot, R.; Tirabosco, R.; Brems, H.; et al. The characteristics of 76 atypical neurofibromas as precursors to neurofibromatosis 1 associated malignant peripheral nerve sheath tumors. Neuro. Oncol. 2018, 20, 818–825. [Google Scholar] [CrossRef] [Green Version]
- Miettinen, M.M.; Antonescu, C.R.; Fletcher, C.D.M.; Bhaumik, S.; Pans, S.; Connor, S.; Miettinen, M.; Sciot, R.; Tirabosco, R.; Brems, H.; et al. Histopathologic evaluation of atypical neurofibromatous tumors and their transformation into malignant peripheral nerve sheath tumor in patients with neurofibromatosis 1-a consensus overview. Hum. Pathol. 2017, 67, 1–10. [Google Scholar] [CrossRef]
- Nielsen, G.P.; Stemmer-Rachamimov, A.O.; Ino, Y.; Moller, M.B.; Rosenberg, A.E.; Louis, D.N. Malignant transformation of neurofibromas in neurofibromatosis 1 is associated with CDKN2A/p16 inactivation. Am. J. Pathol. 1999, 155, 1879–1884. [Google Scholar] [CrossRef] [Green Version]
- Kourea, H.P.; Orlow, I.; Scheithauer, B.W.; Cordon-Cardo, C.; Woodruff, J.M. Deletions of the INK4A gene occur in malignant peripheral nerve sheath tumors but not in neurofibromas. Am. J. Pathol. 1999, 155, 1855–1860. [Google Scholar] [CrossRef] [Green Version]
- Berner, J.M.; Sørlie, T.; Mertens, F.; Henriksen, J.; Saeter, G.; Mandahl, N.; Brøgger, A.; Myklebost, O.; Lothe, R.A. Chromosome band 9p21 is frequently altered in malignant peripheral nerve sheath tumors: Studies of CDKN2A and other genes of the pRB pathway. Genes Chromosomes Cancer 1999, 26, 151–160. [Google Scholar] [CrossRef]
- Menon, A.G.; Anderson, K.M.; Riccardi, V.M.; Chung, R.Y.; Whaley, J.M.; Yandell, D.W.; Farmer, G.E.; Freiman, R.N.; Lee, J.K.; Li, F.P.; et al. Chromosome 17p deletions and p53 gene mutations associated with the formation of malignant neurofibrosarcomas in von Recklinghausen neurofibromatosis. Proc. Natl. Acad. Sci. USA 1990, 87, 5435–5439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Legius, E.; Dierick, H.; Wu, R.; Hall, B.K.; Marynen, P.; Cassiman, J.J.; Glover, T.W. TP53 mutations are frequent in malignant NF1 tumors. Genes Chromosomes Cancer 1994, 10, 250–255. [Google Scholar] [CrossRef] [PubMed]
- Cichowski, K.; Shih, T.S.; Schmitt, E.; Santiago, S.; Reilly, K.; McLaughlin, M.E.; Bronson, R.T.; Jacks, T. Mouse models of tumor development in neurofibromatosis type 1. Science 1999, 286, 2172–2176. [Google Scholar] [CrossRef]
- Hirbe, A.C.; Dahiya, S.; Miller, C.A.; Li, T.; Fulton, R.S.; Zhang, X.; McDonald, S.; DeSchryver, K.; Duncavage, E.J.; Walrath, J.; et al. Whole Exome Sequencing Reveals the Order of Genetic Changes during Malignant Transformation and Metastasis in a Single Patient with NF1-plexiform Neurofibroma. Clin. Cancer Res. 2015, 21, 4201–4211. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Murray, B.; Mo, G.; Shern, J.F. The Role of Polycomb Repressive Complex in Malignant Peripheral Nerve Sheath Tumor. Genes 2020, 11, 287. [Google Scholar] [CrossRef] [Green Version]
- Schindler, G.; Capper, D.; Meyer, J.; Janzarik, W.; Omran, H.; Herold-Mende, C.; Schmieder, K.; Wesseling, P.; Mawrin, C.; Hasselblatt, M.; et al. Analysis of BRAF V600E mutation in 1,320 nervous system tumors reveals high mutation frequencies in pleomorphic xanthoastrocytoma, ganglioglioma and extra-cerebellar pilocytic astrocytoma. Acta Neuropathol. 2011, 121, 397–405. [Google Scholar] [CrossRef]
- Je, E.M.; An, C.H.; Yoo, N.J.; Lee, S.H. Mutational analysis of PIK3CA, JAK2, BRAF, FOXL2, IDH1, AKT1 and EZH2 oncogenes in sarcomas. APMIS 2012, 120, 635–639. [Google Scholar] [CrossRef]
- Peacock, J.D.; Pridgeon, M.G.; Tovar, E.A.; Essenburg, C.J.; Bowman, M.; Madaj, Z.; Koeman, J.; Boguslawski, E.A.; Grit, J.; Dodd, R.D.; et al. Genomic Status of MET Potentiates Sensitivity to MET and MEK Inhibition in NF1-Related Malignant Peripheral Nerve Sheath Tumors. Cancer Res. 2018, 78, 3672–3687. [Google Scholar] [CrossRef] [Green Version]
- Du, X.; Yang, J.; Ylipää, A.; Zhu, Z. Genomic amplification and high expression of EGFR are key targetable oncogenic events in malignant peripheral nerve sheath tumor. J. Hematol. Oncol. 2013, 6, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holtkamp, N.; Malzer, E.; Zietsch, J.; Okuducu, A.F.; Mucha, J.; Mawrin, C.; Mautner, V.F.; Schildhaus, H.U.; von Deimling, A. EGFR and erbB2 in malignant peripheral nerve sheath tumors and implications for targeted therapy. Neuro. Oncol. 2008, 10, 946–957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perrone, F.; Da Riva, L.; Orsenigo, M.; Losa, M.; Jocollè, G.; Millefanti, C.; Pastore, E.; Gronchi, A.; Pierotti, M.A.; Pilotti, S. PDGFRA, PDGFRB, EGFR, and downstream signaling activation in malignant peripheral nerve sheath tumor. Neuro. Oncol. 2009, 11, 725–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, A.V.; Da Riva, L.; Orsenigo, M.; Rizvi, T.A.; Ecsedy, J.A.; Qian, M.G.; Aronow, B.J.; Perentesis, J.P.; Serra, E.; Cripe, T.P.; et al. Ras-driven transcriptome analysis identifies aurora kinase A as a potential malignant peripheral nerve sheath tumor therapeutic target. Clin. Cancer Res. 2012, 18, 5020–5030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirbe, A.C.; Kaushal, M.; Sharma, M.K.; Dahiya, S.; Pekmezci, M.; Perry, A.; Gutmann, D.H. Clinical genomic profiling identifies TYK2 mutation and overexpression in patients with neurofibromatosis type 1-associated malignant peripheral nerve sheath tumors. Cancer 2017, 123, 1194–1201. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, F.J.; Graham, M.K.; Brosnan-Cashman, J.A.; Cashman, J.A.; Barber, J.R.; Davis, C.; Vizcaino, M.A.; Palsgrove, D.N.; Giannini, C.; Pekmezci, M.; et al. Telomere alterations in neurofibromatosis type 1-associated solid tumors. Acta Neuropathol. Commun. 2019, 7, 139. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.M.N.; Deng, Y.; Wang, J.; Zhao, C.; Wang, J.; Rao, R.; Xu, L.; Zhou, W.; Choi, K.; Rizvi, T.A.; et al. Programming of Schwann Cells by Lats1/2-TAZ/YAP Signaling Drives Malignant Peripheral Nerve Sheath Tumorigenesis. Cancer Cell 2018, 33, 292–308.e7. [Google Scholar] [CrossRef] [Green Version]
- Hirbe, A.C.; Pekmezci, M.; Dahiya, S.; Apicelli, A.J.; Van Tine, B.A.; Perry, A.; Gutmann, D.H. BRAFV600E mutation in sporadic and neurofibromatosis type 1-related malignant peripheral nerve sheath tumors. Neuro. Oncol. 2014, 16, 466–467. [Google Scholar] [CrossRef] [Green Version]
- Serrano, C.; Simonetti, S.; Hernández-Losa, J.; Valverde, C.; Carrato, C.; Bagué, S.; Orellana, R.; Somoza, R.; Moliné, T.; Carles, J.; et al. BRAF V600E and KRAS G12S mutations in peripheral nerve sheath tumours. Histopathology 2013, 62, 499–504. [Google Scholar] [CrossRef]
- DeClue, J.E.; Heffelfinger, S.; Benvenuto, G.; Ling, B.; Li, S.; Rui, W.; Vass, W.C.; Viskochil, D.; Ratner, N. Epidermal growth factor receptor expression in neurofibromatosis type 1-related tumors and NF1 animal models. J. Clin. Investig. 2000, 105, 1233–1241. [Google Scholar] [CrossRef] [Green Version]
- Torres, K.E.; Zhu, Q.S.; Bill, K.; Lopez, G.; Ghadimi, M.P.; Xie, X.; Young, E.D.; Liu, J.; Nguyen, T.; Bolshakov, S.; et al. Activated MET is a molecular prognosticator and potential therapeutic target for malignant peripheral nerve sheath tumors. Clin. Cancer Res. 2011, 17, 3943–3955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohan, P.; Castellsague, J.; Jiang, J.; Allen, K.; Chen, H.; Nemirovsky, O.; Spyra, M.; Hu, K.; Kluwe, L.; Pujana, M.A.; et al. Genomic imbalance of HMMR/RHAMM regulates the sensitivity and response of malignant peripheral nerve sheath tumour cells to aurora kinase inhibition. Oncotarget 2013, 4, 80–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, W.; Godec, A.; Zhang, X.; Zhu, C.; Shao, J.; Tao, Y.; Bu, X.; Hirbe, A.C. TYK2 promotes malignant peripheral nerve sheath tumor progression through inhibition of cell death. Cancer Med. 2019, 8, 5232–5241. [Google Scholar] [CrossRef]
- Lu, H.C.; Eulo, V.; Apicelli, A.J.; Pekmezci, M.; Tao, Y.; Luo, J.; Hirbe, A.C.; Dahiya, S. Aberrant ATRX protein expression is associated with poor overall survival in NF1-MPNST. Oncotarget 2018, 9, 23018–23028. [Google Scholar] [CrossRef] [Green Version]
- Jessen, W.J.; Miller, S.J.; Jousma, E.; Wu, J.; Rizvi, T.A.; Brundage, M.E.; Eaves, D.; Widemann, B.; Kim, M.O.; Dombi, E.; et al. MEK inhibition exhibits efficacy in human and mouse neurofibromatosis tumors. J. Clin. Investig. 2013, 123, 340–347. [Google Scholar] [CrossRef]
- Kolberg, M.; Høland, M.; Lind, G.E.; Agesen, T.H.; Skotheim, R.I.; Hall, K.S.; Mandahl, N.; Smeland, S.; Mertens, F.; Davidson, B.; et al. Protein expression of BIRC5, TK1, and TOP2A in malignant peripheral nerve sheath tumours—A prognostic test after surgical resection. Mol. Oncol. 2015, 9, 1129–1139. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Mo, J.; Brosseau, J.P.; Shipman, T.; Wang, Y.; Liao, C.P.; Cooper, J.M.; Allaway, R.J.; Gosline, S.; Guinney, J.; et al. Spatiotemporal Loss of NF1 in Schwann Cell Lineage Leads to Different Types of Cutaneous Neurofibroma Susceptible to Modification by the Hippo Pathway. Cancer Discov. 2019, 9, 114–129. [Google Scholar] [CrossRef] [Green Version]
- Kohlmeyer, J.L.; Kaemmer, C.A.; Pulliam, C.; Maharjan, C.K.; Samayoa, A.M.; Major, H.J.; Cornick, K.E.; Knepper-Adrian, V.; Khanna, R.; Sieren, J.C.; et al. RABL6A is an essential driver of MPNSTs that negatively regulates the RB1 pathway and sensitizes tumor cells to CDK4/6 inhibitors. Clin. Cancer Res. 2020, 26, 2997–3011. [Google Scholar] [CrossRef] [Green Version]
- Brossier, N.M.; Prechtl, A.M.; Longo, J.F.; Barnes, S.; Wilson, L.S.; Byer, S.J.; Brosius, S.N.; Carroll, S.L. Classic Ras Proteins Promote Proliferation and Survival via Distinct Phosphoproteome Alterations in Neurofibromin-Null Malignant Peripheral Nerve Sheath Tumor Cells. J. Neuropathol. Exp. Neurol. 2015, 74, 568–586. [Google Scholar] [CrossRef] [Green Version]
- Nix, J.S.; Haffner, M.C.; Ahsan, S.; Hicks, J.; De Marzo, A.M.; Blakeley, J.; Raabe, E.H.; Rodriguez, F.J. Malignant Peripheral Nerve Sheath Tumors Show Decreased Global DNA Methylation. J. Neuropathol. Exp. Neurol. 2018, 77, 958–963. [Google Scholar] [CrossRef]
- Bradtmoller, M.; Hartmann, C.; Zietsch, J.; Jäschke, S.; Mautner, V.F.; Kurtz, A.; Park, S.J.; Baier, M.; Harder, A.; Reuss, D.; et al. Impaired Pten expression in human malignant peripheral nerve sheath tumours. PLoS ONE 2012, 7, e47595. [Google Scholar] [CrossRef] [PubMed]
- Wojcik, J.B.; Marchione, D.M.; Sidoli, S.; Djedid, A.; Lisby, A.; Majewski, J.; Garcia, B.A. Epigenomic Reordering Induced by Polycomb Loss Drives Oncogenesis but Leads to Therapeutic Vulnerabilities in Malignant Peripheral Nerve Sheath Tumors. Cancer Res. 2019, 79, 3205–3219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malone, C.F.; Fromm, J.A.; Maertens, O.; DeRaedt, T.; Ingraham, R.; Cichowski, K. Defining key signaling nodes and therapeutic biomarkers in NF1-mutant cancers. Cancer Discov. 2014, 4, 1062–1073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheikh, T.N.; Patwardhan, P.P.; Cremers, S.; Schwartz, G.K. Targeted inhibition of glutaminase as a potential new approach for the treatment of NF1 associated soft tissue malignancies. Oncotarget 2017, 8, 94054–94068. [Google Scholar] [CrossRef] [Green Version]
- Lemberg, K.M.; Zhao, L.; Wu, Y.; Veeravalli, V.; Alt, J.; Aguilar, J.; Dash, R.P.; Lam, J.; Tenora, L.; Rodriguez, C.; et al. The novel glutamine antagonist prodrug JHU395 has antitumor activity in malignant peripheral nerve sheath tumor. Mol. Cancer Ther. 2019, 19, 397–408. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, H.G. Vemurafenib treatment of BRAF V600E-mutated malignant peripheral nerve sheath tumor. J. Natl. Compr. Cancer Netw. 2013, 11, 1466–1470. [Google Scholar] [CrossRef] [Green Version]


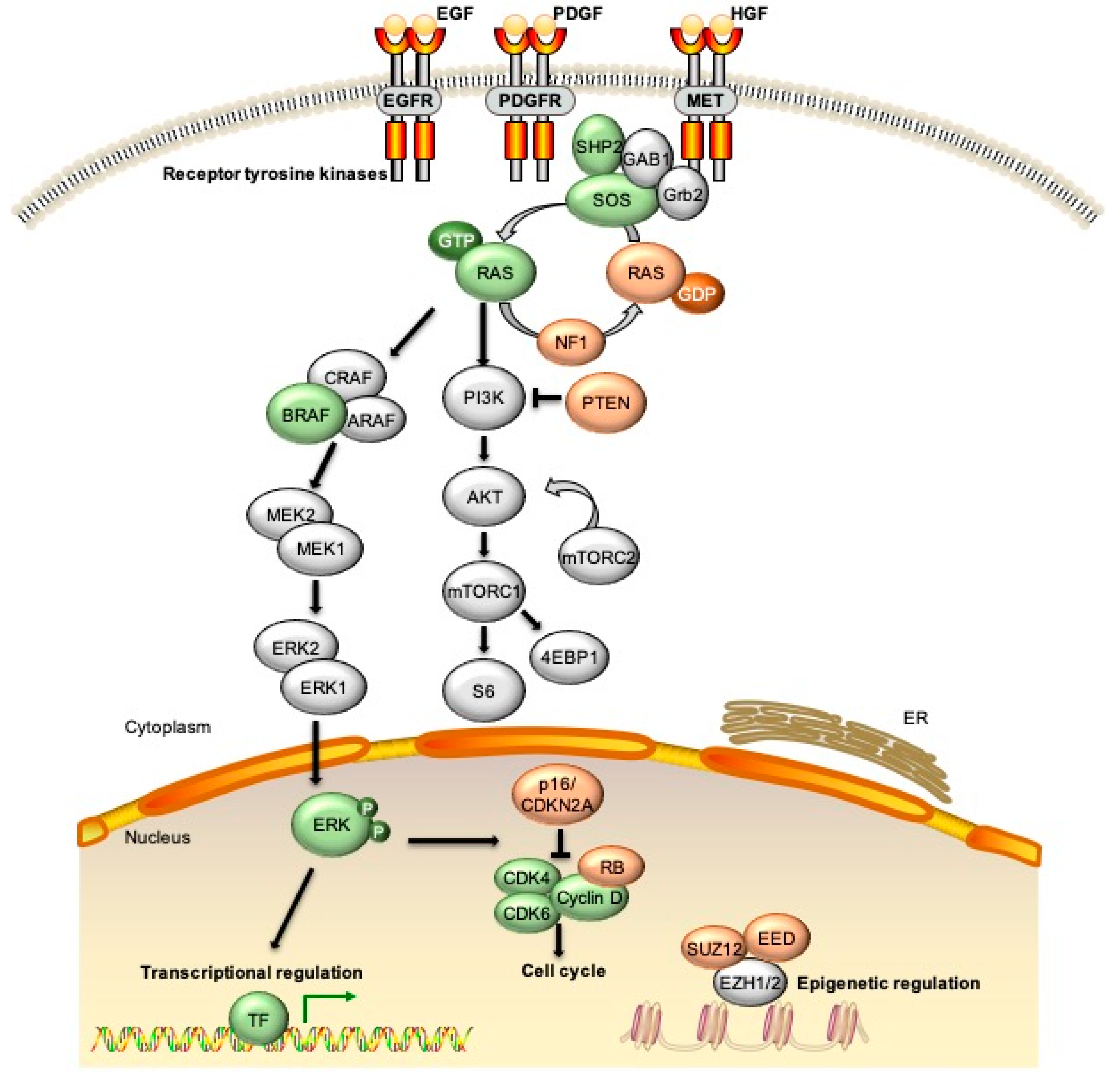{kind=link}
| Ref. | Study Author Year | Description | n Total MPNST (n NF1 Associated) n (Other Specimen Types) | NF1 | CDKN2A | TP53 | EED | SUZ12 | Notes |
|---|---|---|---|---|---|---|---|---|---|
| [38] | Mantripragada, 2008 | Targeted seq, aCGH | 35 (35) 16 pNF 8 cNF | 71% | 39% | 17% | NR | NR | |
| [39] | Verdijk, 2010 | Targeted seq | 88 (26) | NR | ND | 17/72 | ND | ND | 36% of TP53 mutations detected were from NF1 patients |
| [40] | Yang, 2011 | aCGH | 51 (16) | ~30% | 65% | ~30% | NR | NR | |
| [41] | DeRaedt, 2014 | Targeted seq, aCGH | 51 (51) | 51/51 | NR | NR | 15/51 | 32/51 | |
| [42] | Zhang, 2014 | WGS (5), WES (3), Targeted seq (42) | 50 (39) 11 (paired neurofibroma) | 22/50 | 1/8 | 1/8 | 1/50 | 16/50 | |
| [43] | Lee, 2014 | WES (15), SNP, targeted (37) | 52 (27) 7 neurofibromas | 45/52 | 42/52 | 23/52 | 19/52 | 25/52 | RNAseq analysis of MPNST with PRC2 loss vs. intact PRC2 demonstrates enrichment of genes associated with development and morphogenesis |
| [44] | Sohier, 2017 | Exome seq, aCGH | 8 (8) 1 pNF 7 cNF | 8/8 | 5/8 | 1/8 | 2/8 | 7/8 | No TP53 point mutations identified |
| [45] | Brohl, 2017 | WES + SNP | 5 (4) + 7 TCGA cases (6) | 11/12 | 7/12 | 6/12 | 4/12 | 5/12 | 5/12 MPNST contain somatic Ras-pathway activating mutation |
| [46] | Zehir, 2017 | IMPACT NGS | 11 | 2/11 | 6/11 | NR | 1/11 | 2/11 | Data accessible through cBioPortal |
| [47] | Kaplan, 2018 | Foundation Medicine NGS 2014–2016 | 186 (clinical data NR) | 102 of 186 | 57% overall (71% NF1-altered, 80% BRAF altered, 34% non-NF1/non-BRAF altered) | 32% of NF1 14% of non-NF1 | 8% of NF1-altered, 13% of BRAF-altered, 3% of non–NF1/non–BRAF-altered | 20% of NF1-altered, 13% of BRAF-altered, 9% of non–NF1/non–BRAF-altered | Data reported as % of NF1/BRAF cohorts rather than absolute numbers |
| [48] | Pemov, 2019 | NF1 deep sequencing (4); WES (3); CNV (28) | 31 (4) 16 ANF | 4/4; 10/28 (Loss, CNV) | 4/4; 20/28 (Loss, CNV) | 0/3 (WES); 10/28 (Loss, CNV) | 1/3 (WES); 10/28 (Loss, CNV) | 1/3 (WES); 9/28 (Loss, CNV) | RNAseq reported for ANF and 4 MPNST |
| [49] | Pollard, 2020 | WES | 1 (1) 7 pNF 13 cNF | 1 | 0/1 | 0/1 | 0/1 | 1/1 | RNAseq on cNF, pNF, and MPNST samples from 23 patients |
| Gene | Description | n | NF1 | Altered | Details | Study | Ref. |
|---|---|---|---|---|---|---|---|
| BRAF | Targeted seq | 18 | NR | 0 | 18 MPNST out of 1,320 nervous system tumors | Schindler, 2011 | [68] |
| Targeted seq | 47 | 25 | 1 | 1/1 N581S | Bottillo, 2009 | [32] | |
| Targeted seq | 24 | NR | 0 | 0/24 MPNST with BRAF exon 15 mutation | Je, 2012 | [69] | |
| Foundation NGS | 186 | 102 | 10 | 5 of 10 BRAF V600E; 9 of 10 pathogenic; 1 of 10 VUS 47% with alteration in >/=1 non-NF1/non-BRAF gene in the RAS/RAF pathway (ERBB2, ERBB3, ERBB4, KRAS, MET, HRAS, MAP2K1, MAP2K2, NRAS). 7% with alteration in RTK (e.g., KIT/PDGFR/FGFR1)—some likely pathogenic 70% with alteration in DNA repair genes (ATM, BARD1, BRCA1/2, FANCx, PBRM1, CHEK2, MSH2, MSH3, MSH6, NBN, PBRM1, POLE, RAD51, RAD51C) | Kaplan, 2018 | [47] | |
| MET | WES | 1 | 1 | 1 (amplified) | Single patient longitudinal sampling (pre/post treatment, recurrence, mets) Copy number alterations in HGF, EGFR | Peacock, 2018 | [70] |
| Targeted seq, aCGH | 35 | 25% (amplified) | HGF, EGR, PDGFRA amplifications in 25-29% samples | Mantripragada, 2008 | [38] | ||
| EGFR | aCGH | 51 | 37% (19/51) | At least one EGFR pathway gene was altered in 84% of samples, including GRB2, HRAS, MAPK1, STAT1, and others. | Du, 2013 | [71] | |
| Targeted gene sequencing | 37 | 29/37 | 28% gain | Direct sequencing of EGFR exons 18–24 | Holtkamp, 2008 | [72] | |
| Targeted gene sequencing and FISH | 27 | 14 of 25 pts | 14 of 23 (copy number gain) | Direct sequencing of EGFR exons 18–21 | Perrone, 2009 | [73] | |
| IGF1R | aCGH | 51 | 16 | 24% (amplified) | >/= 1 gene in IGFR1 pathway altered in 82% cases | Yang, 2011 | [40] |
| AURKA | SNP array, qPCR | 13 | NR | 8 | 1/8 neurofibromas also with AURKA locus copy number increase | Patel, 2012 | [74] |
| TYK2 | 7 | 7 | 2 | Tyrosine kinase 2, activates STAT signaling and promotes cancer cell survival | Hirbe, 2017 | [75] | |
| ATRX | NGS clinical genomic profiling | 7 | 7 | NR | Hirbe, 2017 | [75] | |
| NGS | 4 | 4 | 2 | Of 3 ALT-positive MPNST, 2 had ATRX mutations. One ALT positive MPNST had RECQL4 variant. | Rodriguez, 2019 | [76] | |
| WGS, WES | 8 | 5 | 1 | Additional chromatin organization-related genes: EZH2, CHD4, and AEBP2 mutations n = 1 tumor each; RBBP7 mutation in n = 2 tumors. | Zhang, 2014 | [42] | |
| KDM2B | Exome seq, aCGH | 8 | 8 | 1 | Jumonji histone lysine demethylase; identified in single patient MPNST lacking SUZ12 or EED mutation | Sohier, 2017 | [44] |
| LATS2 | aCGH | 51 | 16 | NR (copy number loss in ~25%) | Copy number gains and losses in HIPPO effector loci (TAZ, CTGF, BIRC5) and HIPPO inhibitory loci (LATS2, AMOTL2) graphically illustrated. Same dataset as Yang et al. Clin Can Res 2011. | Wu, 2018 | [77] |
| HMMR/RHAMM | aCGH | 35 | 71% | 46% | Deletions in hyaluronan binding protein may affect signaling through ERK or AURAK | Mantripragada, 2008 | [38] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lemberg, K.M.; Wang, J.; Pratilas, C.A. From Genes to -Omics: The Evolving Molecular Landscape of Malignant Peripheral Nerve Sheath Tumor. Genes 2020, 11, 691. https://doi.org/10.3390/genes11060691
Lemberg KM, Wang J, Pratilas CA. From Genes to -Omics: The Evolving Molecular Landscape of Malignant Peripheral Nerve Sheath Tumor. Genes. 2020; 11(6):691. https://doi.org/10.3390/genes11060691
Chicago/Turabian StyleLemberg, Kathryn M., Jiawan Wang, and Christine A. Pratilas. 2020. "From Genes to -Omics: The Evolving Molecular Landscape of Malignant Peripheral Nerve Sheath Tumor" Genes 11, no. 6: 691. https://doi.org/10.3390/genes11060691
APA StyleLemberg, K. M., Wang, J., & Pratilas, C. A. (2020). From Genes to -Omics: The Evolving Molecular Landscape of Malignant Peripheral Nerve Sheath Tumor. Genes, 11(6), 691. https://doi.org/10.3390/genes11060691