De Novo Assembly-Based Analysis of RPGR Exon ORF15 in an Indigenous African Cohort Overcomes Limitations of a Standard Next-Generation Sequencing (NGS) Data Analysis Pipeline

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient Cohort
2.2. DNA Extraction
2.3. ORF15 NGS Amplification and Sequencing
2.4. NGS Data Analysis
2.5. Confirmation by Sanger Sequencing
3. Results
3.1. NGS PCR and Sequencing Efficiency
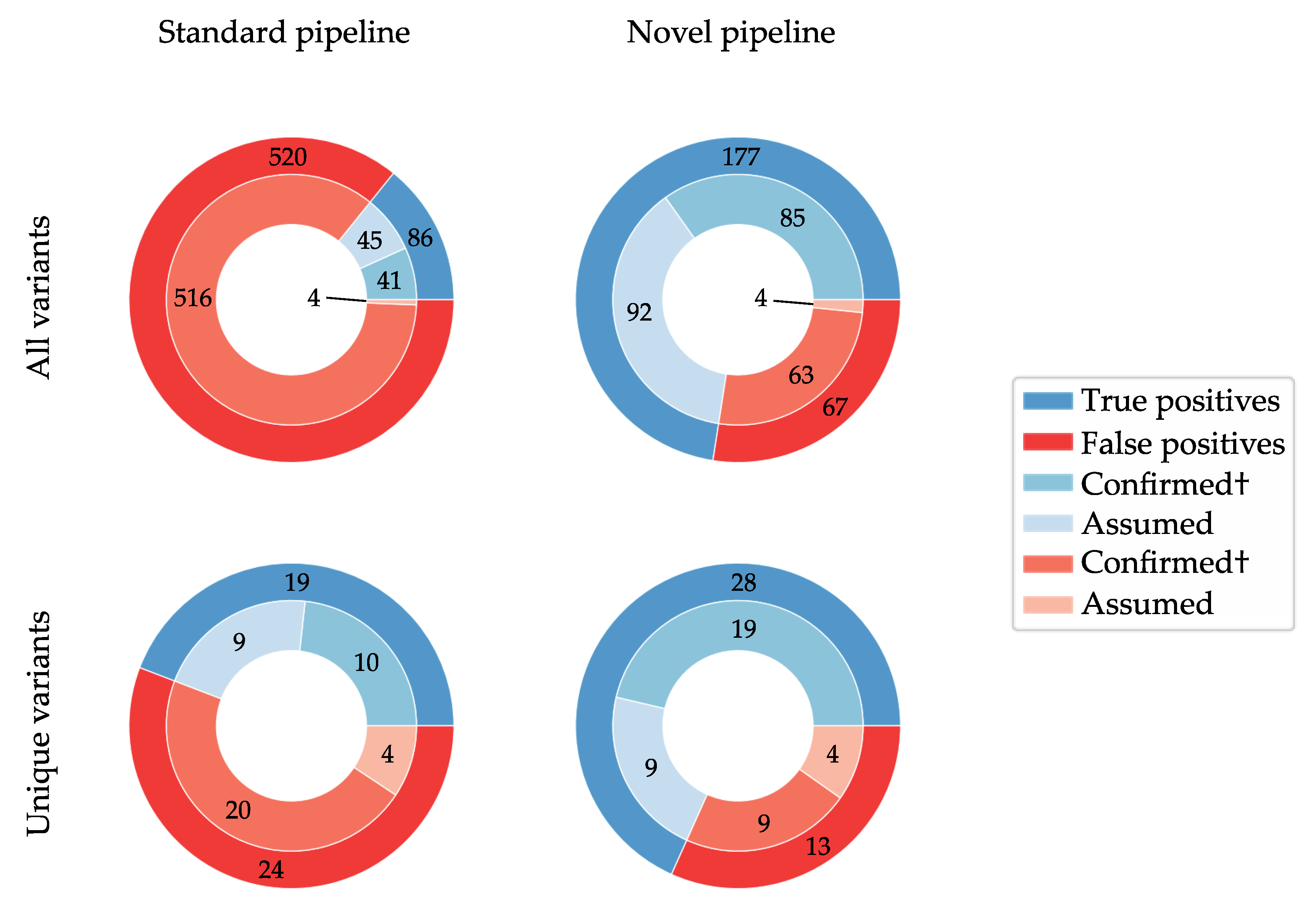
3.2. De Novo Assembly Improves Diagnostic Sensitivity
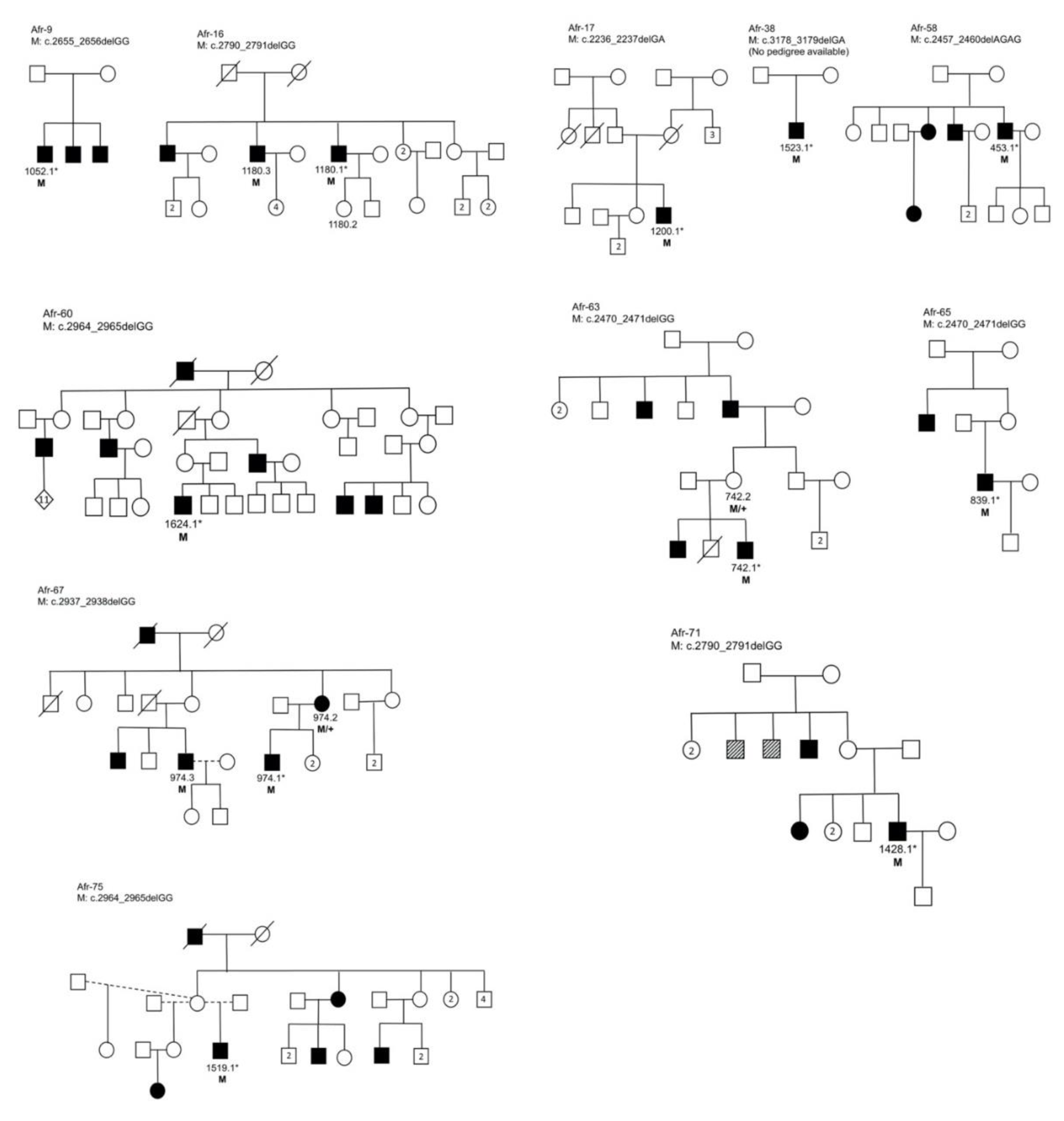
3.3. Segregation Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Berger, W.; Kloeckener-Gruissem, B.; Neidhardt, J. The molecular basis of human retinal and vitreoretinal diseases. Prog. Retin. Eye Res. 2010, 29, 335–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrari, S.; Di Iorio, E.; Barbaro, V.; Ponzin, D.; Sorrentino, F.S.; Parmeggiani, F. Retinitis pigmentosa: Genes and disease mechanisms. Curr. Genomics 2011, 12, 238–249. [Google Scholar] [PubMed]
- Ayuso, C.; Millan, J.M. Retinitis pigmentosa and allied conditions today: A paradigm of translational research. Genome Med. 2010, 2, 34–44. [Google Scholar] [CrossRef]
- Branham, K.; Othman, M.; Brumm, M.; Karoukis, A.J.; Atmaca-Sonmez, P.; Yashar, B.M.; Schwartz, S.B.; Stover, N.B.; Trzupek, K.; Wheaton, D.; et al. Mutations in RPGR and RP2 account for 15% of males with simplex retinal degenerative disease. Investig. Ophthalmol. Vis. Sci. 2012, 53, 8232–8237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porter, L.F.; Black, G.C.M. Personalized ophthalmology. Clin. Genet. 2014, 86, 1–11. [Google Scholar] [CrossRef]
- Koenekoop, R.K.; Loyer, M.; Hand, C.K.; Al Mahdi, H.; Dembinska, O.; Beneish, R.; Racine, J.; Rouleau, G.A. Novel RPGR mutations with distinct retinitis pigmentosa phenotypes in French-Canadian families. Am. J. Ophthalmol. 2003, 136, 678–687. [Google Scholar] [CrossRef]
- Breuer, D.K.; Yashar, B.M.; Filippova, E.; Hiriyanna, S.; Lyons, R.H.; Mears, A.J.; Asaye, B.; Acar, C.; Vervoort, R.; Wright, A.F.; et al. A comprehensive mutation analysis of RP2 and RPGR in a North American cohort of families with X-linked retinitis pigmentosa. Am. J. Hum. Genet. 2002, 70, 1545–1554. [Google Scholar] [CrossRef] [Green Version]
- Schwahn, U.; Lenzner, S.; Dong, J.; Feil, S.; Hinzmann, B.; Van Duijnhoven, G.; Kirschner, R.; Hemberger, M.; Bergen, A.A.B.; Rosenberg, T.; et al. Positional cloning of the gene for X-linked retinitis pigmentosa 2. Nat. Genet. 1998, 19, 327–332. [Google Scholar] [CrossRef]
- Roepman, R.; van Duijnhoven, G.; Rosenberg, T.; Pinckers, A.J.; Bleeker-Wagemakers, L.M.; Bergen, A.A.; Post, J.; Beck, A.; Reinhardt, R.; Ropers, H.H.; et al. Positional cloning of the gene for X-linked retinitis pigmentosa 3: Homology with the guanine-nucleotide-exchange factor RCC1. Hum. Mol. Genet. 1996, 5, 1035–1041. [Google Scholar] [CrossRef] [Green Version]
- Webb, T.R.; Parfitt, D.A.; Gardner, J.C.; Martinez, A.; Bevilacqua, D.; Davidson, A.E.; Zito, I.; Thiselton, D.L.; Ressa, J.H.C.C.; Apergi, M.; et al. Deep intronic mutation in OFD1, identified by targeted genomic next-generation sequencing, causes a severe form of X-linked retinitis pigmentosa (RP23). Hum. Mol. Genet. 2012, 21, 3647–3654. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.; Berg, J.S.; Milko, L.; Crooks, K.; Lu, M.; Bizon, C.; Owen, P.; Wilhelmsen, K.C.; Weck, K.E.; Evans, J.P.; et al. High diagnostic yield of whole exome sequencing in participants with retinal dystrophies in a clinical ophthalmology setting. Am. J. Ophthalmol. 2015, 160, 354–363.e9. [Google Scholar] [CrossRef] [Green Version]
- Ratnapriya, R.; Swaroop, A. Genetic architecture of retinal and macular degenerative diseases: The promise and challenges of next-generation sequencing. Genome Med. 2013, 5, 84–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, D.M.; Khanna, H.; Atmaca-Sonmez, P.; Sieving, P.A.; Branham, K.; Othman, M.; Swaroop, A.; Daiger, S.P.; Heckenlively, J.R. Long-term follow-up of a family with dominant X-linked retinitis pigmentosa. Eye 2010, 24, 764–774. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, A.; Maruiwa, F.; Hayakawa, M.; Kanai, A.; Vervoort, R.; Wright, A.F.; Yamada, K.; Niikawa, N.; Naōi, N. Three novel mutations of the RPGR gene exon ORF15 in three Japanese families with X-linked retinitis pigmentosa. Am. J. Med. Genet. 2001, 104, 232–238. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, U.; Warburg, M.; Jørgensen, A.L. X-inactivation pattern in carriers of X-linked retinitis pigmentosa: A valuable means of prognostic evaluation? Hum. Genet. 1993, 92, 359–363. [Google Scholar] [CrossRef]
- Grover, S.; Fishman, G.A.; Anderson, R.J.; Lindeman, M. A longitudinal study of visual function in carriers of X-linked recessive retinitis pigmentosa. Ophthalmology 2000, 6420, 386–396. [Google Scholar] [CrossRef]
- Comander, J.; Weigel-DiFranco, C.; Sandberg, M.A.; Berson, E.L. Visual function in carriers of X-linked retinitis pigmentosa. Ophthalmology 2015, 122, 1899–1906. [Google Scholar] [CrossRef] [Green Version]
- Pyo Park, S.; Hwan Hong, I.; Tsang, S.H.; Chang, S. Cellular imaging demonstrates genetic mosaicism in heterozygous carriers of an X-linked ciliopathy gene. Eur. J. Hum. Genet 2013, 21, 1240–1248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pomares, E.; Riera, M.; Castro-Navarro, J.; Andrés-Gutiérrez, A.; Gonzàlez-Duarte, R.; Marfany, G. Identification of an intronic single-point mutation in RP2 as the cause of semidominant X-linked retinitis pigmentosa. Investig. Ophthalmol. Vis. Sci. 2009, 50, 5107–5114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banin, E.; Mizrahi-Meissonnier, L.; Neis, R.; Silverstein, S.; Magyar, I.; Abeliovich, D.; Roepman, R.; Berger, W.; Rosenberg, T.; Sharon, D. A non-ancestral RPGR missense mutation in families with either recessive or semi-dominant X-linked retinitis pigmentosa. Am. J. Med. Genet. A 2007, 143A, 1150–1158. [Google Scholar] [CrossRef] [PubMed]
- Souied, E.; Segues, B.; Ghazi, I.; Rozet, J.M.; Chatelin, S.; Gerber, S.; Perrault, I.; Michel-Awad, A.; Briard, M.L.; Plessis, G.; et al. Severe manifestations in carrier females in X linked retinitis pigmentosa. J. Med. Genet 1997, 34, 793–797. [Google Scholar] [CrossRef] [Green Version]
- Vervoort, R.; Lennon, A.; Bird, A.C.; Tulloch, B.; Axton, R.; Miano, M.G.; Meindl, A.; Meitinger, T.; Ciccodicola, A.; Wright, A.F. Mutational hot spot within a new RPGR exon in X-linked retinitis pigmentosa. Nat. Genet. 2000, 25, 462–466. [Google Scholar] [CrossRef]
- Kirschner, R.; Rosenberg, T.; Schultz-Heienbrok, R.; Lenzner, S.; Feil, S.; Roepman, R.; Cremers, F.P.M.; Ropers, H.H.; Berger, W. RPGR transcription studies in mouse and human tissues reveal a retina-specific isoform that is disrupted in a patient with X-linked retinitis pigmentosa. Hum. Mol. Genet. 1999, 8, 1571–1578. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.F.; Wu, J.; Lv, J.N.; Zhang, X.; Jin, Z.B. Identification of false-negative mutations missed by next-generation sequencing in retinitis pigmentosa patients: A complementary approach to clinical genetic diagnostic testing. Genet. Med. 2015, 17, 307–311. [Google Scholar] [CrossRef]
- Roberts, L.; Ratnapriya, R.; du Plessis, M.; Chaitankar, V.; Ramesar, R.S.; Swaroop, A. Molecular diagnosis of inherited retinal diseases in indigenous African populations by whole-exome sequencing. Investig. Ophthalmol. Vis. Sci. 2016, 57, 6374–6381. [Google Scholar] [CrossRef] [Green Version]
- Schuster, S.C.; Miller, W.; Ratan, A.; Tomsho, L.P.; Giardine, B.; Kasson, L.R.; Harris, R.S.; Petersen, D.C.; Zhao, F.; Qi, J.; et al. Complete Khoisan and Bantu genomes from southern Africa. Nature 2010, 463, 943–947. [Google Scholar] [CrossRef] [Green Version]
- May, A.; Hazelhurst, S.; Li, Y.; Norris, S.A.; Govind, N.; Tikly, M.; Hon, C.; Johnson, K.J.; Hartmann, N.; Staedtler, F.; et al. Genetic diversity in black South Africans from Soweto. BMC Genom. 2013, 14, 644–655. [Google Scholar] [CrossRef] [Green Version]
- Henn, B.M.; Botigué, L.R.; Peischl, S.; Dupanloup, I.; Lipatov, M.; Maples, B.K.; Martin, A.R.; Musharoff, S.; Cann, H.; Snyder, M.P.; et al. Distance from sub-Saharan Africa predicts mutational load in diverse human genomes. Proc. Natl. Acad. Sci. USA 2016, 113, E440–E449. [Google Scholar] [CrossRef] [Green Version]
- Chimusa, E.R.; Meintjies, A.; Tchanga, M.; Mulder, N.; Seoighe, C.; Soodyall, H.; Ramesar, R. A Genomic portrait of haplotype diversity and signatures of selection in indigenous Southern African populations. PLoS Genet. 2015, 11, e1005052. [Google Scholar] [CrossRef] [Green Version]
- Choudhury, A.; Ramsay, M.; Hazelhurst, S.; Aron, S.; Bardien, S.; Botha, G.; Chimusa, E.R.; Christoffels, A.; Gamieldien, J.; Sefid-Dashti, M.J.; et al. Whole-genome sequencing for an enhanced understanding of genetic variation among South Africans. Nat. Commun. 2017, 8, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Sherman, R.M.; Forman, J.; Antonescu, V.; Puiu, D.; Daya, M.; Rafaels, N.; Boorgula, M.P.; Chavan, S.; Vergara, C.; Ortega, V.E.; et al. Assembly of a pan-genome from deep sequencing of 910 humans of African descent. Nat. Genet. 2019, 51, 30–35. [Google Scholar] [CrossRef]
- Pierrache, L.H.M.; Kimchi, A.; Ratnapriya, R.; Roberts, L.; Astuti, G.D.N.; Obolensky, A.; Beryozkin, A.; Tjon-Fo-Sang, M.J.H.; Schuil, J.; Klaver, C.C.W.; et al. Whole-exome sequencing identifies biallelic IDH3A variants as a cause of retinitis pigmentosa accompanied by pseudocoloboma. Ophthalmology 2017, 124, 992–1003. [Google Scholar] [CrossRef] [PubMed]
- Shu, X.; Black, G.C.; Rice, J.M.; Hart-Holden, N.; Jones, A.; O’Grady, A.; Ramsden, S.; Wright, A.F. RPGR mutation analysis and disease: An update. Hum. Mutat. 2007, 28, 322–328. [Google Scholar] [CrossRef]
- García-Hoyos, M.; Garcia-Sandoval, B.; Cantalapiedra, D.; Riveiro, R.; Lorda-Sánchez, I.; Trujillo-Tiebas, M.J.; Rodriguez de Alba, M.; Millan, J.M.; Baiget, M.; Ramos, C.; et al. Mutational screening of the RP2 and RPGR genes in Spanish families with X-linked retinitis pigmentosa. Investig. Ophthalmol. Vis. Sci. 2006, 47, 3777–3782. [Google Scholar] [CrossRef] [PubMed]
- Oliver, G.R.; Hart, S.N.; Klee, E.W. Bioinformatics for clinical next generation sequencing. Clin. Chem. 2015, 61, 124–135. [Google Scholar] [CrossRef]
- Miller, S.A.; Dykes, D.D.; Polesky, H.F. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988, 16, 1215. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Tang, J.; Feng, Y.; Xu, M.; Chen, R.; Zou, X.; Sui, R.; Chang, E.Y.; Lewis, R.A.; Zhang, V.W.; et al. Improved diagnosis of inherited retinal dystrophies by high-fidelity PCR of ORF15 followed by next-generation sequencing. J. Mol. Diagnostics 2016, 18, 817–824. [Google Scholar] [CrossRef] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Page, A.J.; Taylor, B.; Delaney, A.J.; Soares, J.; Seemann, T.; Keane, J.A.; Harris, S.R. SNP-sites: Rapid efficient extraction of SNPs from multi-FASTA alignments. Microb. Genom. 2016, 2, e000056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. bioRxiv 2019, 531210. [Google Scholar] [CrossRef] [Green Version]
- Shu, X.; McDowall, E.; Brown, A.F.; Wright, A.F. The human retinitis pigmentosa GTPase regulator gene variant database. Hum. Mutat. 2008, 29, 605–608. [Google Scholar] [CrossRef] [PubMed]
- Carss, K.; Arno, G.; Erwood, M.; Stephens, J.; Sanchis-Juan, A.; Hull, S.; Megy, K.; Grozeva, D.; Dewhurst, E.; Malka, S.; et al. Comprehensive rare variant analysis via whole-genome sequencing to determine the molecular pathology of inherited retinal disease. Am. J. Hum. Genet. 2017, 100, 75–90. [Google Scholar] [CrossRef] [Green Version]
- Bravo-Gil, N.; Méndez-Vidal, C.; Romero-Pérez, L.; González-Del Pozo, M.; Rodríguez-De La Ruá, E.; Dopazo, J.; Borrego, S.; Antinõlo, G. Improving the management of inherited retinal dystrophies by targeted sequencing of a population-specific gene panel. Sci. Rep. 2016, 6, 1–10. [Google Scholar] [CrossRef]
- Neidhardt, J.; Glaus, E.; Lorenz, B.; Netzer, C.; Li, Y.; Schambeck, M.; Wittmer, M.; Feil, S.; Kirschner-Schwabe, R.; Rosenberg, T.; et al. Identification of novel mutations in X-linked retinitis pigmentosa families and implications for diagnostic testing. Mol. Vis. 2008, 14, 1081–1092. [Google Scholar]
- Hall, T.A.T. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Wildeman, M.; van Ophuizen, E.; den Dunnen, J.T.; Taschner, P.E.M. Improving sequence variant descriptions in mutation databases and literature using the Mutalyzer sequence variation nomenclature checker. Hum. Mutat. 2008, 29, 6–13. [Google Scholar] [CrossRef]
- Sharon, D.; Sandberg, M.A.; Rabe, V.W.; Stillberger, M.; Dryja, T.P.; Berson, E.L. RP2 and RPGR mutations and clinical correlations in patients with X-linked retinitis pigmentosa. Am. J. Hum. Genet. 2003, 73, 1131–1146. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| cNomen (NM_001034853.2) | pNomen | # of Samples | gnomAD (%) | rsID | Pathogenicity | Std Pipeline | Sanger | First Report |
|---|---|---|---|---|---|---|---|---|
| c.*125delA | 52 | 31.2591 | rs35637775 | Benign | No | |||
| c.3407G>A | p.Gly1136Asp | 2 | 0.2171 | rs150960964 | Benign | Yes | ||
| c.3396C>T | p.= | 4 | 16.1297 | rs12687163 | Benign | Yes | ||
| c.3264G>A | p.= | 14 | 7.7524 | rs78736275 | Benign | Yes | ||
| c.3219C>T | p.= | 14 | 7.7934 | rs111787313 | Benign | Yes | Yes † | |
| c.3178_3179del | p.Glu1060ArgfsTer18 | 1 | 0.0011 | rs771214648 | Pathogenic | Yes | Yes | 1 |
| c.3108_3122del | p.Glu1038_Glu1042del | 1 | 0.2447 | rs774012136 | Benign | Yes | ||
| c.3074T>G | p.Val1025Gly | 1 | 0.0169 | rs773842474 | Likely benign | Yes | No | |
| c.3062T>G | p.Val1021Gly | 1 | 0.8368* | rs144299434 | Likely benign | Yes | No | |
| c.2964_2965del | p.Glu989GlyfsTer89 | 2 | NA | Pathogenic | Yes | Yes | 1 | |
| c.2919_2939dup | p.Gly977_Glu983dup | 13 | 1.0322* | rs772859148 | VUS | No | Yes † | |
| c.2937_2938del | p.Glu980GlyfsTer98 | 1 | NA | Pathogenic | Yes | Yes | 2 | |
| c.2863T>G | p.Trp955Gly | 47 | 19.7002 | rs765224300 | Benign | Yes | No † | |
| c.2820_2840dup | p.Asp943_Glu949dup | 5 | 3.3006 | rs764268405 | Benign | No | Yes | |
| c.2829T>G | p.Asp943Glu | 1 | 1.4892 | rs201655057 | Likely benign | Yes | ||
| c.2829T>A | p.Asp943Glu | 1 | 0.133 | rs201655057 | Likely benign | Yes | ||
| c.2820A>G | p.= | 1 | NA | VUS | No | No | ||
| c.2790_2791del | p.Glu931GlyfsTer147 | 2 | NA | Pathogenic | Yes | Yes | 3 | |
| c.2778_2786del | p.Glu927_Glu929del | 1 | NA | VUS | Yes | |||
| c.2758A>G | p.Lys920Glu | 4 | 0.0548 | rs1241379586 | Likely benign | Yes | No † | |
| c.2705G>A | p.Gly902Glu | 1 | NA | Likely benign | Yes | |||
| c.2667_2669del | p.Glu890del | 1 | 1.6129 | rs1263452259 | Benign | Yes | ||
| c.2660_2661insGGAAGAGGAGGAAGGAGAAGGGGAGGGAGAAGAGGAAGGAGAAGGGGAGGG | p.Glu890_Gly891insGluGlyGluGlyGluGlyGluGluGluGlyGluGlyGluGlyGluGluGlu | 1 | NA | VUS | No | Yes | ||
| c.2655_2656del | p.Glu886GlyfsTer192 | 1 | NA | Pathogenic | Yes | Yes | 4 | |
| c.2639A>G | p.Glu880Gly | 1 | NA | Likely benign | Yes | Yes | ||
| c.2633_2634delinsAA | p.Gly878Glu | 1 | NA | Likely benign | Yes | Yes | ||
| c.2618_2632dup | p.Glu873_Glu877dup | 5 | NA | VUS | No | Yes † | ||
| c.2606_2632dup | p.Glu869_Glu877dup | 6 | NA | rs769216492 | VUS | No | Yes † | |
| c.2569A>G | p.Lys857Glu | 6 | 0.981 | rs1250133030 | Likely benign | Yes | No † | |
| c.2541_2561del | p.Glu850_Gly856del | 12 | 2.0962 | rs886038384 | Likely benign | No | Yes † | |
| c.2514G>A | p.= | 1 | NA | VUS | Yes | |||
| c.2499T>G | p.= | 1 | 0.0406 | rs752979508 | Likely benign | Yes | No | |
| c.2470_2471del | p.Gly824ArgfsTer10 | 2 | NA | Pathogenic | Yes | Yes | This study | |
| c.2457_2460del | p.Glu820ArgfsTer268 | 1 | NA | Pathogenic | Yes | Yes | This study | |
| c.2341G>A | p.Ala781Thr | 2 | 12.1386 | rs5917557 | Benign | Yes | ||
| c.2236_2237del | p.Glu746ArgfsTer23 | 1 | NA | Pathogenic | Yes | Yes | 5 | |
| c.2223G>A | p.= | 15 | 10.3568 | rs147619484 | Benign | Yes | Yes † | |
| c.2057T>A | p.Met686Lys | 1 | 0.449 | rs151247357 | Likely benign | Yes | ||
| c.1961G>A | p.Arg654Lys | 1 | NA | Likely benign | Yes | No | ||
| c.1933G>A | p.Gly645Arg | 1 | NA | Likely benign | Yes | No | ||
| c.1754-103C>T | 15 | 10.1753 | rs41303691 | Benign | Yes |
| cNomen (NM_001034853.2) | pNomen | # of Samples | Het. (Het. Males) | Hom./Hemi. | Novel Pipeline (# of Samples) | Call Quality | Sanger |
|---|---|---|---|---|---|---|---|
| c.3407G>A | p.Gly1136Asp | 3 | 2 (0) | 1 | Yes (2) | High | |
| c.3396C>T | p.= | 5 | 2 (0) | 3 | Yes (4) | High | |
| c.3356G>A | p.Arg1119Lys | 1 | 1 (1) | 0 | No | Low | |
| c.3264G>A | p.= | 15 | 4 (0) | 11 | Yes (14) | High | |
| c.3219C>T | p.= | 15 | 4 (0) | 11 | Yes (14) | High | Yes † |
| c.3178_3179del | p.Glu1060ArgfsTer18 | 1 | 0 (0) | 1 | Yes (1) | High | Yes |
| c.3108_3122del | p.Glu1038_Glu1042del | 1 | 0 (0) | 1 | Yes (1) | High | |
| c.3074T>G | p.Val1025Gly | 80 | 80 (64) | 0 | Yes (1) | Low | No † |
| c.3062T>G | p.Val1021Gly | 74 | 74 (58) | 0 | Yes (1) | Low | No † |
| c.2964_2965del | p.Glu989GlyfsTer89 | 2 | 2 (2) | 0 | Yes (2) | High | Yes |
| c.2937_2938del | p.Glu980GlyfsTer98 | 1 | 1 (1) | 0 | Yes (1) | High | Yes |
| c.2895G>A | p.= | 8 | 8 (7) | 0 | No | Low | No † |
| c.2876A>G | p.Glu959Gly | 1 | 1 (1) | 0 | No | Low | No |
| c.2863T>G | p.Trp955Gly | 81 | 81 (65) | 0 | Yes (47) | Low | No † |
| c.2847A>G | p.= | 1 | 1 (1) | 0 | No | Low | No |
| c.2829T>G | p.Asp943Glu | 50 | 50 (44) | 0 | Yes (1) | Low | No † |
| c.2829T>A | p.Asp943Glu | 30 | 40 (23) | 0 | Yes (1) | Low | No † |
| c.2790_2791del | p.Glu931GlyfsTer147 | 2 | 2 (2) | 0 | Yes (2) | High | Yes |
| c.2778_2786del | p.Glu927_Glu929del | 1 | 0 (0) | 1 | Yes (1) | High | |
| c.2784A>G | p.= | 1 | 1 (1) | 0 | No | Low | No |
| c.2758A>G | p.Lys920Glu | 75 | 75 (59) | 0 | Yes (4) | Low | No † |
| c.2705G>A | p.Gly902Glu | 1 | 1 (1) | 0 | Yes (1) | High | |
| c.2667_2669del | p.Glu890del | 2 | 1 (0) | 1 | Yes (1) | High | |
| c.2655_2656del | p.Glu886GlyfsTer192 | 1 | 1 (1) | 0 | Yes (1) | High | Yes |
| c.2639A>G | p.Glu880Gly | 6 | 6 (6) | 0 | Yes (1) | Low | No † |
| c.2634G>A | p.= | 6 | 6 (6) | 0 | Yes (1) | Low | No † |
| c.2633G>A | p.Gly878Glu | 6 | 6 (6) | 0 | Yes (1) | Low | No † |
| c.2589A>G | p.= | 7 | 7 (6) | 0 | No | Low | No † |
| c.2569A>G | p.Lys857Glu | 11 | 11 (9) | 0 | Yes (6) | Low | No † |
| c.2531A>G | p.Glu844Gly | 1 | 1 (0) | 0 | No | Low | |
| c.2517A>G | p.= | 11 | 11 (10) | 0 | No | Low | No † |
| c.2514G>A | p.= | 1 | 1 (1) | 0 | Yes (1) | High | |
| c.2499T>G | p.= | 65 | 65 (54) | 0 | Yes (1) | Low | No † |
| c.2470_2471del | p.Gly824ArgfsTer10 | 2 | 0 (0) | 2 | Yes (2) | High | Yes |
| c.2457_2460del | p.Glu820ArgfsTer268 | 1 | 0 (0) | 1 | Yes (1) | High | Yes |
| c.2341G>A | p.Ala781Thr | 2 | 1 (0) | 1 | Yes (2) | High | |
| c.2236_2237del | p.Glu746ArgfsTer23 | 1 | 0 (0) | 1 | Yes (1) | High | Yes |
| c.2223G>A | p.= | 15 | 3 (0) | 12 | Yes (15) | High | Yes † |
| c.2057T>A | p.Met686Lys | 1 | 0 (0) | 1 | Yes (1) | High | |
| c.1961G>A | p.Arg654Lys | 1 | 1 (1) | 0 | Yes (1) | High | No |
| c.1933G>A | p.Gly645Arg | 1 | 1 (1) | 0 | Yes (1) | High | No |
| c.1885G>A | p.Asp629Asn | 1 | 1 (1) | 0 | No | Low | No |
| c.1754-103C>T | 15 | 10.1753 | rs41303691 | Benign | Yes |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maggi, J.; Roberts, L.; Koller, S.; Rebello, G.; Berger, W.; Ramesar, R. De Novo Assembly-Based Analysis of RPGR Exon ORF15 in an Indigenous African Cohort Overcomes Limitations of a Standard Next-Generation Sequencing (NGS) Data Analysis Pipeline. Genes 2020, 11, 800. https://doi.org/10.3390/genes11070800
Maggi J, Roberts L, Koller S, Rebello G, Berger W, Ramesar R. De Novo Assembly-Based Analysis of RPGR Exon ORF15 in an Indigenous African Cohort Overcomes Limitations of a Standard Next-Generation Sequencing (NGS) Data Analysis Pipeline. Genes. 2020; 11(7):800. https://doi.org/10.3390/genes11070800
Chicago/Turabian StyleMaggi, Jordi, Lisa Roberts, Samuel Koller, George Rebello, Wolfgang Berger, and Rajkumar Ramesar. 2020. "De Novo Assembly-Based Analysis of RPGR Exon ORF15 in an Indigenous African Cohort Overcomes Limitations of a Standard Next-Generation Sequencing (NGS) Data Analysis Pipeline" Genes 11, no. 7: 800. https://doi.org/10.3390/genes11070800
APA StyleMaggi, J., Roberts, L., Koller, S., Rebello, G., Berger, W., & Ramesar, R. (2020). De Novo Assembly-Based Analysis of RPGR Exon ORF15 in an Indigenous African Cohort Overcomes Limitations of a Standard Next-Generation Sequencing (NGS) Data Analysis Pipeline. Genes, 11(7), 800. https://doi.org/10.3390/genes11070800