Preferentially Paternal Origin of De Novo 11p13 Chromosome Deletions Revealed in Patients with Congenital Aniridia and WAGR Syndrome

 ,
,
Abstract
:1. Introduction
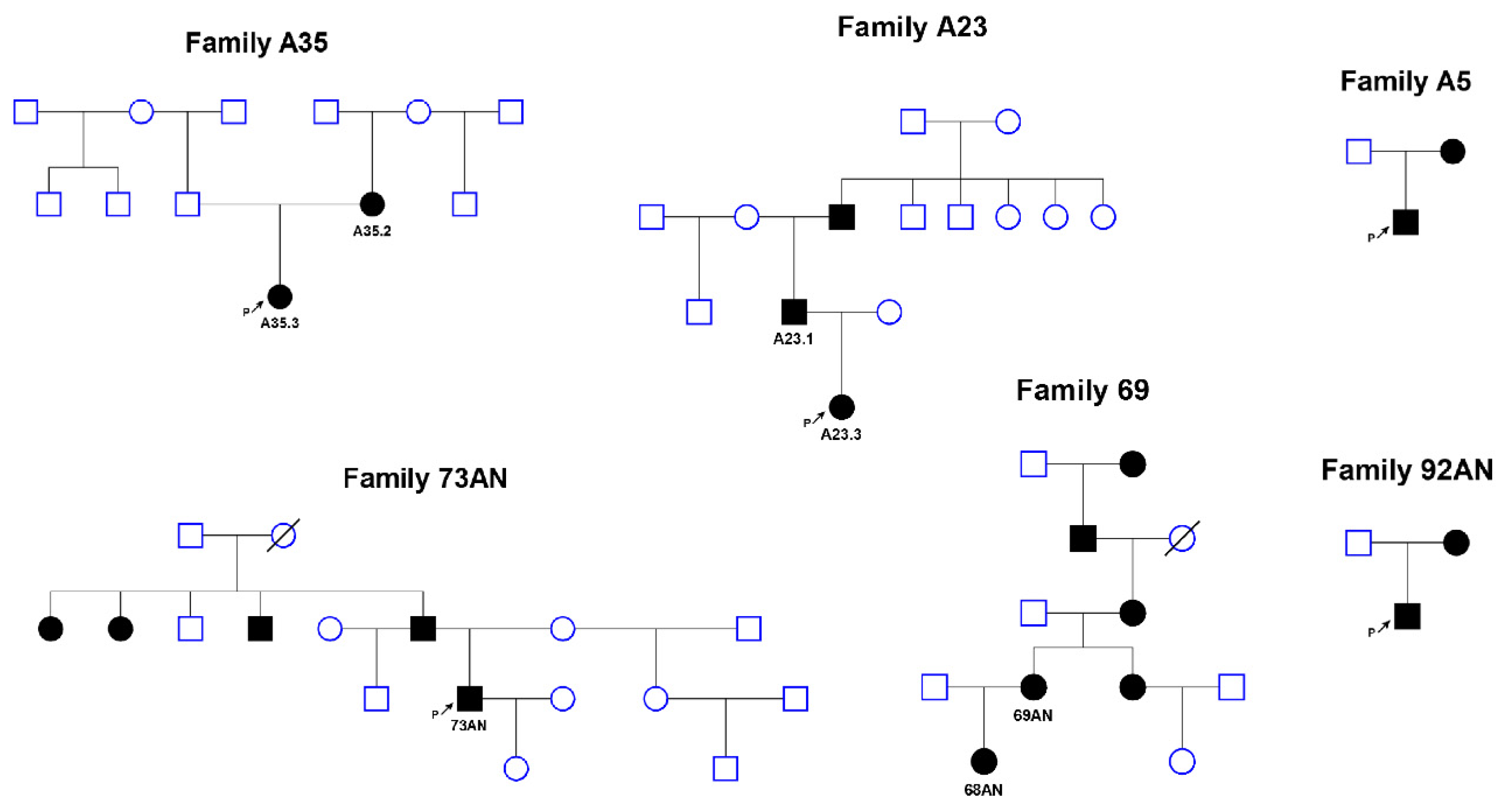
2. Materials and Methods
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Käsmann-Kellner, B.; Seitz, B. Aniridia syndrome: Clinical findings, problematic courses and suggestions for optimization of care (“aniridia guide”). Ophthalmologe 2014, 111, 1145–1156. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.T.; Kim, H.; Kim, H. PAX6 aniridia syndrome. Curr. Opin. Ophthalmol. 2017, 28, 436–447. [Google Scholar] [CrossRef] [PubMed]
- Grønskov, K.; Olsen, J.H.; Sand, A.; Pedersen, W.; Carlsen, N.; Jylling, A.M.B.; Lyngbye, T.; Brøndum-Nielsen, K.; Rosenberg, T. Population-based risk estimates of Wilms tumor in sporadic aniridia. A comprehensive mutation screening procedure of PAX6 identifies 80% of mutations in aniridia. Hum. Genet. 2001, 109, 11–18. [Google Scholar] [CrossRef]
- Marakhonov, A.V.; A Vasilyeva, T.; A Voskresenskaya, A.; Sukhanova, N.V.; Kadyshev, V.V.; I Kutsev, S.; A Zinchenko, R. LMO2 gene deletions significantly worsen the prognosis of Wilms’ tumor development in patients with WAGR syndrome. Hum. Mol. Genet. 2019, 28, 3323–3326. [Google Scholar] [CrossRef]
- Robinson, D.O.; Howarth, R.J.; Williamson, K.A.; Van Heyningen, V.; Beal, S.J.; Crolla, J.A. Genetic analysis of chromosome 11p13 and thePAX6 gene in a series of 125 cases referred with aniridia. Am. J. Med. Genet. Part A 2008, 146, 558–569. [Google Scholar] [CrossRef]
- Vasilyeva, T.A.; Marakhonov, A.V.; Voskresenskaya, A.A.; Kadyshev, V.V.; Käsmann-Kellner, B.; Sukhanova, N.V.; Katargina, L.A.; Kutsev, S.I.; Zinchenko, R.A. Analysis of genotype–phenotype correlations in PAX6-associated aniridia. J. Med. Genet. 2020. [Google Scholar] [CrossRef]
- Jimeno, S.; Carvajal, R.-P.; Huertas, P. The role of RNA and RNA-related proteins in the regulation of DNA double strand break repair pathway choice. DNA Repair 2019, 81, 102662. [Google Scholar] [CrossRef]
- Karlsson, K.H.; Stenerlöw, B. Extensive ssDNA end formation at DNA double-strand breaks in non-homologous end-joining deficient cells during the S phase. BMC Mol. Boil. 2007, 8, 97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jabbari, K.; Wirtz, J.; Rauscher, M.; Wiehe, T. A common genomic code for chromatin architecture and recombination landscape. PLoS ONE 2019, 14, e0213278. [Google Scholar] [CrossRef] [Green Version]
- Gothe, H.J.; Bouwman, B.A.M.; Gusmao, E.G.; Piccinno, R.; Petrosino, G.; Sayols, S.; Drechsel, O.; Minneker, V.; Josipovic, N.; Mizi, A.; et al. Spatial Chromosome Folding and Active Transcription Drive DNA Fragility and Formation of Oncogenic MLL Translocations. Mol. Cell 2019, 75, 267–283.e12. [Google Scholar] [CrossRef]
- Alexander, J.L.; Orr-Weaver, T.L. Replication fork instability and the consequences of fork collisions from rereplication. Genes Dev. 2016, 30, 2241–2252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tock, A.J.; Henderson, I.R. Hotspots for Initiation of Meiotic Recombination. Front. Genet. 2018, 9, 521. [Google Scholar] [CrossRef] [PubMed]
- Vasilyeva, T.A.; Voskresenskaya, A.A.; Käsmann-Kellner, B.; Khlebnikova, O.V.; Pozdeyeva, N.; Bayazutdinova, G.M.; Kutsev, S.I.; Ginter, E.K.; Semina, E.V.; Marakhonov, A.V.; et al. Molecular analysis of patients with aniridia in Russian Federation broadens the spectrum of PAX6 mutations. Clin. Genet. 2017, 92, 639–644. [Google Scholar] [CrossRef]
- Pellestor, F.; Anahory, T.; Lefort, G.; Puechberty, J.; Liehr, T.; Hédon, B.; Sarda, P. Complex chromosomal rearrangements: origin and meiotic behavior. Hum. Reprod. Updat. 2011, 17, 476–494. [Google Scholar] [CrossRef] [PubMed]
- Hehir-Kwa, J.Y.; Rodríguez-Santiago, B.; E Vissers, L.; De Leeuw, N.; Pfundt, R.; Buitelaar, J.K.; A Pérez-Jurado, L.; A Veltman, J. De novo copy number variants associated with intellectual disability have a paternal origin and age bias. J. Med. Genet. 2011, 48, 776–778. [Google Scholar] [CrossRef] [PubMed]
- Roychowdhury, T.; Abyzov, A. Chromatin organization modulates the origin of heritable structural variations in human genome. Nucleic Acids Res. 2019, 47, 2766–2777. [Google Scholar] [CrossRef] [Green Version]
- Steinmann, K.; Cooper, D.N.; Kluwe, L.; Chuzhanova, N.; Senger, C.; Serra, E.; Lazaro, C.; Gilaberte, M.; Wimmer, K.; Mautner, V.-F.; et al. Type 2 NF1 Deletions Are Highly Unusual by Virtue of the Absence of Nonallelic Homologous Recombination Hotspots and an Apparent Preference for Female Mitotic Recombination. Am. J. Hum. Genet. 2007, 81, 1201–1220. [Google Scholar] [CrossRef] [Green Version]
- Van Binsbergen, E. Origins and Breakpoint Analyses of Copy Number Variations: Up Close and Personal. Cytogenet. Genome Res. 2011, 135, 271–276. [Google Scholar] [CrossRef]
- Huff, V.; Meadows, A.; Riccardi, V.M.; Strong, L.C.; Saunders, G.F. Parental origin of de novo constitutional deletions of chromosomal band 11p13. Am. J. Hum. Genet. 1990, 47, 155–160. [Google Scholar]
- Parvanov, E.; Petkov, P.M.; Paigen, K. Prdm9 Controls Activation of Mammalian Recombination Hotspots. Science 2009, 327, 835. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, K.; Yoshida, K.; Matsui, Y. A histone H3 methyltransferase controls epigenetic events required for meiotic prophase. Nature 2005, 438, 374–378. [Google Scholar] [CrossRef] [PubMed]
- Broman, K.W.; Murray, J.C.; Sheffield, V.C.; White, R.L.; Weber, J.L. Comprehensive Human Genetic Maps: Individual and Sex-Specific Variation in Recombination. Am. J. Hum. Genet. 1998, 63, 861–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.-L.; Gokcumen, O. Fine-Scale Characterization of Genomic Structural Variation in the Human Genome Reveals Adaptive and Biomedically Relevant Hotspots. Genome Boil. Evol. 2019, 11, 1136–1151. [Google Scholar] [CrossRef] [PubMed]
- Gold, H.B.; Jung, Y.H.; Corces, V.G. Not just heads and tails: The complexity of the sperm epigenome. J. Boil. Chem. 2018, 293, 13815–13820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rathke, C.; Baarends, W.M.; Awe, S.; Renkawitz-Pohl, R. Chromatin dynamics during spermiogenesis. Biochim. Biophys. Acta (BBA) Bioenerg. 2014, 1839, 155–168. [Google Scholar] [CrossRef] [Green Version]
- LeDuc, F.; Maquennehan, V.; Nkoma, G.B.; Boissonneault, G. DNA Damage Response During Chromatin Remodeling in Elongating Spermatids of Mice1. Boil. Reprod. 2008, 78, 324–332. [Google Scholar] [CrossRef]
- Crolla, J.A.; Van Heyningen, V. Frequent Chromosome Aberrations Revealed by Molecular Cytogenetic Studies in Patients with Aniridia. Am. J. Hum. Genet. 2002, 71, 1138–1149. [Google Scholar] [CrossRef] [Green Version]
- Lebedev, I.N.; Sazhenova, Y.A. Epimutations of imprinting genes in the human genome: classification, causes, association with hereditary pathology. Russ. J. Genet. 2008, 44, 1176–1190. [Google Scholar] [CrossRef]
- Zhu, Q.; Stöger, R.; Alberio, R. A Lexicon of DNA Modifications: Their Roles in Embryo Development and the Germline. Front. Cell Dev. Boil. 2018, 6, 24. [Google Scholar] [CrossRef] [Green Version]
- Wossidlo, M.; Arand, J.; Sebastiano, V.; Lepikhov, K.; Boiani, M.; Reinhardt, R.; Scholer, H.; Walter, J. Dynamic link of DNA demethylation, DNA strand breaks and repair in mouse zygotes. EMBO J. 2010, 29, 1877–1888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derijck, A.; Van Der Heijden, G.; Giele, M.; Philippens, M.E.; De Boer, P. DNA double-strand break repair in parental chromatin of mouse zygotes, the first cell cycle as an origin of de novo mutation. Hum. Mol. Genet. 2008, 17, 1922–1937. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Marin, C.; Gosalvez, J.; Roy, R. Types, Causes, Detection and Repair of DNA Fragmentation in Animal and Human Sperm Cells. Int. J. Mol. Sci. 2012, 13, 14026–14052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wyck, S.; Herrera, C.; Requena, C.E.; Bittner, L.; Hajkova, P.; Bollwein, H.; Santoro, R. Oxidative stress in sperm affects the epigenetic reprogramming in early embryonic development. Epigenetics Chromatin 2018, 11, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ladstätter, S.; Tachibana, K. A Surveillance Mechanism Ensures Repair of DNA Lesions during Zygotic Reprogramming. Cell 2016, 167, 1774–1787.e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LeDuc, F.; Nkoma, G.B.; Boissonneault, G. Spermiogenesis and DNA Repair: A Possible Etiology of Human Infertility and Genetic Disorders. Syst. Boil. Reprod. Med. 2008, 54, 3–10. [Google Scholar] [CrossRef]
- Himes, K.P.; Young, A.; Koppes, E.; Stolz, D.B.; Barak, Y.; Sadovsky, Y.; Chaillet, J. Loss of inherited genomic imprints in mice leads to severe disruption in placental lipid metabolism. Placenta 2015, 36, 389–396. [Google Scholar] [CrossRef] [Green Version]
- Ruan, Z.; Zhao, X.; Qin, X.; Luo, C.; Liu, X.; Deng, Y.; Zhu, P.; Li, Z.; Huang, B.; Shi, D.; et al. DNA methylation and expression of imprinted genes are associated with the viability of different sexual cloned buffaloes. Reprod. Domest. Anim. 2017, 53, 203–212. [Google Scholar] [CrossRef] [Green Version]
- Shaw, M.W.; Falls, H.F.; Neel, J.V. Congenital Aniridia. Am. J. Hum. Genet. 1960, 12, 389–415. [Google Scholar]
- Monk, D. Germline-derived DNA methylation and early embryo epigenetic reprogramming: The selected survival of imprints. Int. J. Biochem. Cell Boil. 2015, 67, 128–138. [Google Scholar] [CrossRef]


{kind=link}
| Patient’s ID | Break Points Coordinates According to 11p13 MLPA | Genes Affected by Deletion | Origin 1 |
|---|---|---|---|
| A-25 | chr11:31824328–31832887 | PAX6ex1–PAX6ex5 | nd |
| A-36 | chr11:31671656–32339851 | ELP4ex9–PAX6–RCN1 | pat |
| A-30 | chr11:27679822–33374888 | BNDF−FSHB−DCDC1−ELP4–PAX6−RCN1−WT1−HIPK3 | pat |
| 52.03 | chr11:27679822–35160813 | BNDF−FSHB−DCDC1−ELP4–PAX6−RCN1−WT1−HIPK3−LMO2−EHF−CD44 | pat |
| 20.03 | chr11:30253552–32457265 | FSHB–DCDC1−ELP4−PAX6–RCN1−WT1 | pat |
| 02.12 | chr11:31329311–31671656 | DCDC1−ELP4ex9 | pat |
| 09.03 | chr11:31329311–31671656 | DCDC1−ELP4ex9 | nd |
| 04.14 | chr11:31391209–31838055 | DCDC1ex1−ELP4−PAX6int1 | mat |
| 36.03 | chr11:30253552–32125308 | PAX6ex7−RCN1 | pat |
| A-26 | chr11:31329311–32339851 | DCDC1-ELP–PAX6–RCN1 | pat |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vasilyeva, T.A.; Marakhonov, A.V.; Sukhanova, N.V.; Kutsev, S.I.; Zinchenko, R.A. Preferentially Paternal Origin of De Novo 11p13 Chromosome Deletions Revealed in Patients with Congenital Aniridia and WAGR Syndrome. Genes 2020, 11, 812. https://doi.org/10.3390/genes11070812
Vasilyeva TA, Marakhonov AV, Sukhanova NV, Kutsev SI, Zinchenko RA. Preferentially Paternal Origin of De Novo 11p13 Chromosome Deletions Revealed in Patients with Congenital Aniridia and WAGR Syndrome. Genes. 2020; 11(7):812. https://doi.org/10.3390/genes11070812
Chicago/Turabian StyleVasilyeva, Tatyana A., Andrey V. Marakhonov, Natella V. Sukhanova, Sergey I. Kutsev, and Rena A. Zinchenko. 2020. "Preferentially Paternal Origin of De Novo 11p13 Chromosome Deletions Revealed in Patients with Congenital Aniridia and WAGR Syndrome" Genes 11, no. 7: 812. https://doi.org/10.3390/genes11070812
APA StyleVasilyeva, T. A., Marakhonov, A. V., Sukhanova, N. V., Kutsev, S. I., & Zinchenko, R. A. (2020). Preferentially Paternal Origin of De Novo 11p13 Chromosome Deletions Revealed in Patients with Congenital Aniridia and WAGR Syndrome. Genes, 11(7), 812. https://doi.org/10.3390/genes11070812