Therapeutic Strategies for Duchenne Muscular Dystrophy: An Update


Abstract
:1. Introduction
2. Genetic Pathogenesis of DMD
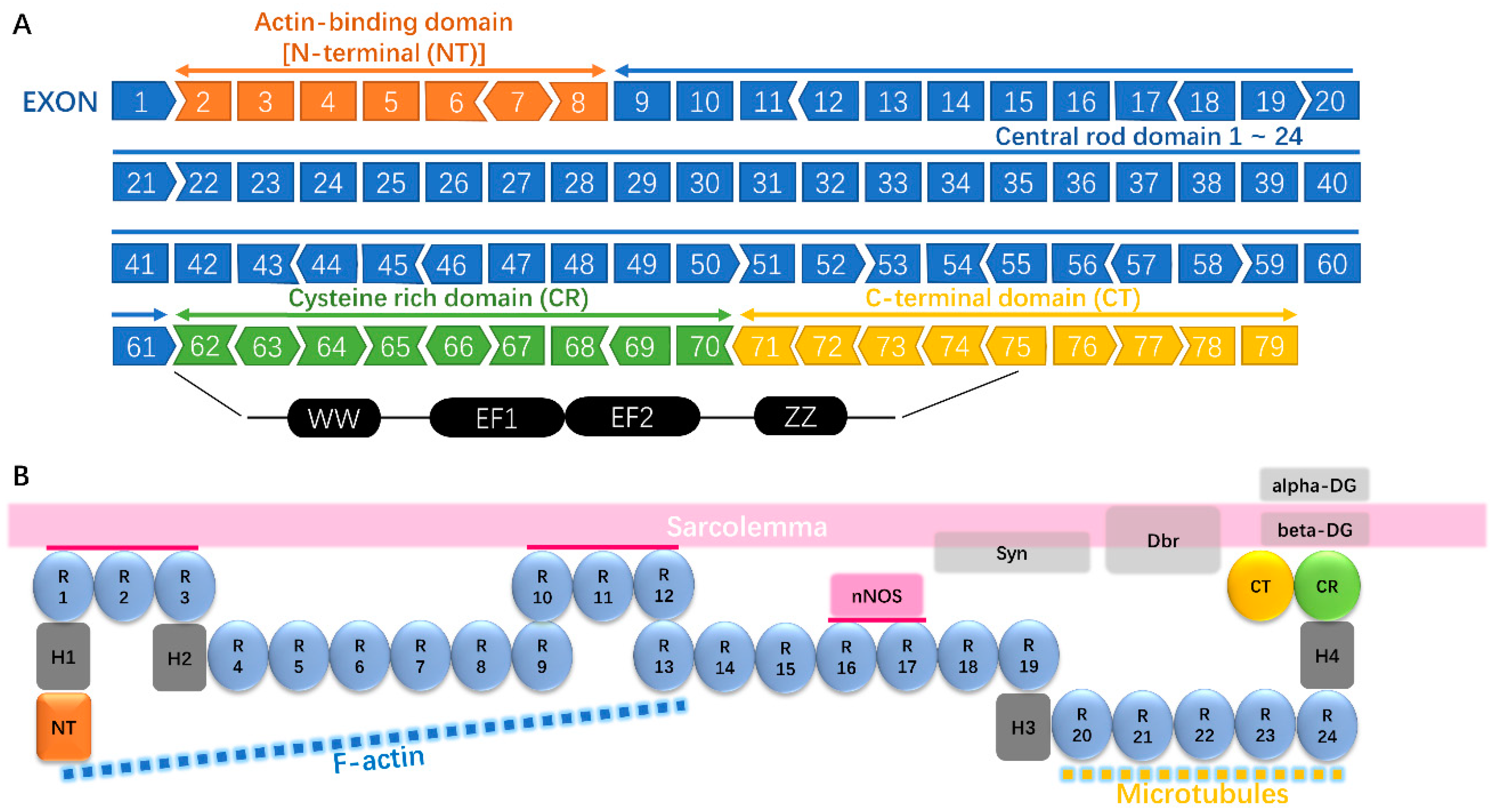
2.1. An Overview of Dystrophin Gene Mutations—Types and Sites
2.2. Correlation between Mutations and Disease Severity
2.3. Diagnosis Techniques Targeting Mutated Exons
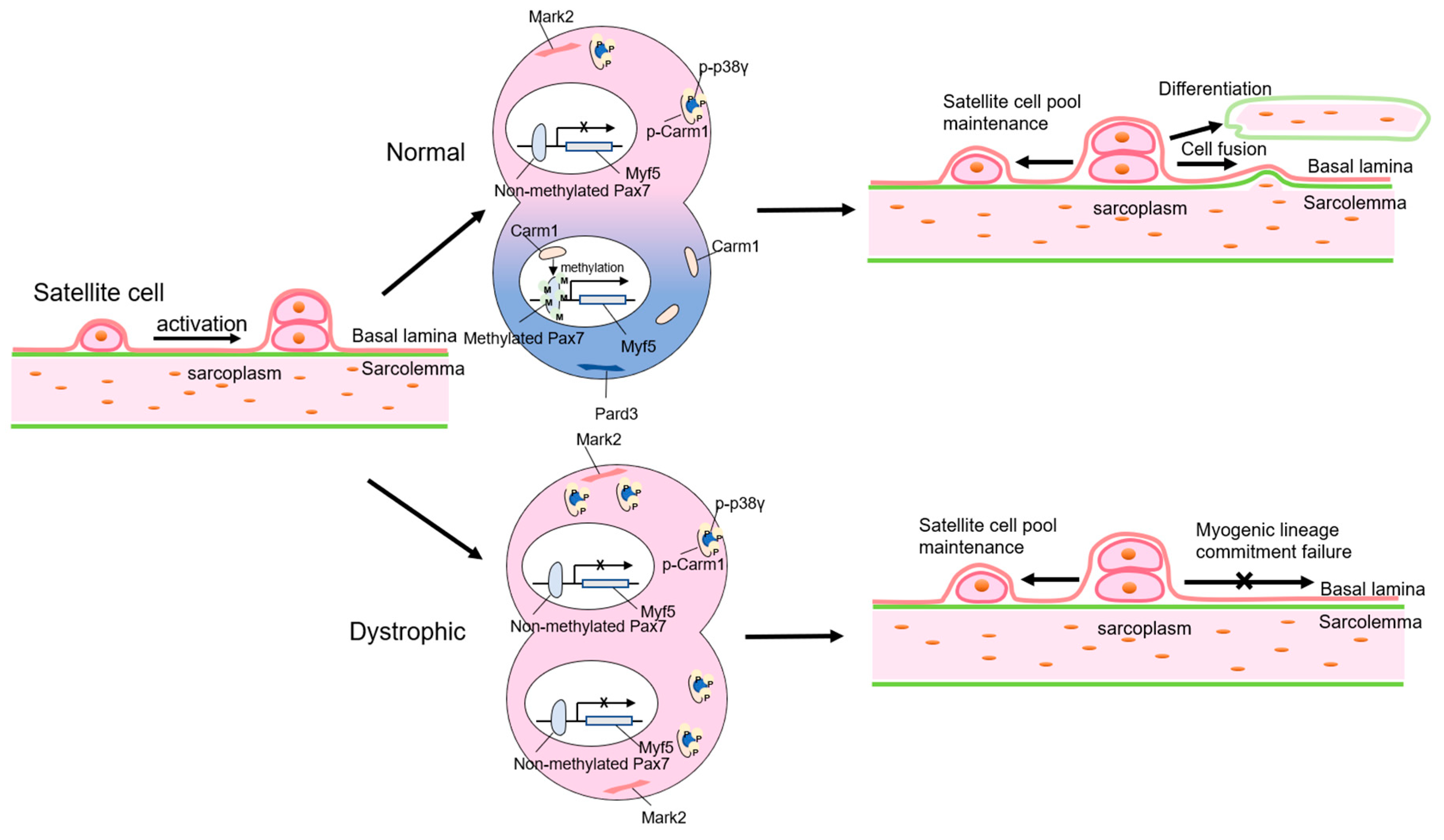
3. Stem Cell-Related DMD Pathogenesis
4. Therapeutic Strategies for DMD
4.1. Read-Through Therapy
4.1.1. Antibiotics and Synthetic Analogues that Mediate Stop-Codon Read-Through
4.1.2. Ataluren-Mediated Stop-Codon Read-Through
4.2. AON-Mediated Exon Skipping Therapy
4.2.1. Phosphorodiamidate Morpholino Oligomer (PMO) Modification
4.2.2. 2′-O-Methyl-Phosphorothioate (2′OMePS) Modification
4.2.3. Peptide-Conjugated PMO (PPMO)
4.2.4. Stereopure AON
4.2.5. Efficacy and Safety of AON-Mediated Exon Skipping
4.3. Vector-Mediated Gene Therapy
4.3.1. AAV-Mediated Mini-/Microdystrophin Transfer
4.3.2. Artificial Chromosome-Mediated Dystrophin Transfer
4.4. CRISPR/Cas9-Mediated Gene Editing
4.4.1. Ex Vivo CRISPR/Cas9 Gene Editing
4.4.2. In Vivo CRISPR/Cas9 Gene Editing
4.5. Exogenous Cell Transplantation
4.6. Level of Functional Dystrophin Required for Clinical Efficacy
5. Discussion and Future Direction
Author Contributions
Funding
Conflicts of Interest
References
- Kolwicz, S.C., Jr.; Hall, J.K.; Moussavi-Harami, F.; Chen, X.; Hauschka, S.D.; Chamberlain, J.S.; Regnier, M.; Odom, G.L. Gene Therapy Rescues Cardiac Dysfunction in Duchenne Muscular Dystrophy Mice by Elevating Cardiomyocyte Deoxy-Adenosine Triphosphate. JACC Basic Transl. Sci. 2019, 4, 778–791. [Google Scholar] [CrossRef] [PubMed]
- Verhaart, I.E.C.; Aartsma-Rus, A. Therapeutic developments for Duchenne muscular dystrophy. Nat. Rev. Neurol. 2019, 15, 373–386. [Google Scholar] [CrossRef] [PubMed]
- Koenig, M.; Monaco, A.P.; Kunkel, L.M. The complete sequence of dystrophin predicts a rod-shaped cytoskeletal protein. Cell 1988, 53, 219–228. [Google Scholar] [CrossRef]
- Salmaninejad, A.; Jafari Abarghan, Y.; Bozorg Qomi, S.; Bayat, H.; Yousefi, M.; Azhdari, S.; Talebi, S.; Mojarrad, M. Common therapeutic advances for Duchenne muscular dystrophy (DMD). Int. J. Neurosci. 2020, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Salmaninejad, A.; Valilou, S.F.; Bayat, H.; Ebadi, N.; Daraei, A.; Yousefi, M.; Nesaei, A.; Mojarrad, M. Duchenne muscular dystrophy: An updated review of common available therapies. Int. J. Neurosci. 2018, 128, 854–864. [Google Scholar] [CrossRef]
- Matre, P.R.; Mu, X.; Wu, J.; Danila, D.; Hall, M.A.; Kolonin, M.G.; Darabi, R.; Huard, J. CRISPR/Cas9-Based Dystrophin Restoration Reveals a Novel Role for Dystrophin in Bioenergetics and Stress Resistance of Muscle Progenitors. Stem Cells 2019, 37, 1615–1628. [Google Scholar] [CrossRef] [Green Version]
- Min, Y.L.; Bassel-Duby, R.; Olson, E.N. CRISPR Correction of Duchenne Muscular Dystrophy. Annu. Rev. Med. 2019, 70, 239–255. [Google Scholar] [CrossRef]
- Bladen, C.L.; Salgado, D.; Monges, S.; Foncuberta, M.E.; Kekou, K.; Kosma, K.; Dawkins, H.; Lamont, L.; Roy, A.J.; Chamova, T.; et al. The TREAT-NMD DMD Global Database: Analysis of More than 7000 Duchenne Muscular Dystrophy Mutations. Hum. Mutat. 2015, 36, 395–402. [Google Scholar] [CrossRef]
- Aartsma-Rus, A.; Van Deutekom, J.C.T.; Fokkema, I.F.; Van Ommen, G.-J.B.; Den Dunnen, J.T. Entries in the Leiden Duchenne muscular dystrophy mutation database: An overview of mutation types and paradoxical cases that confirm the reading-frame rule. Muscle Nerve 2006, 34, 135–144. [Google Scholar] [CrossRef]
- Koenig, M.; Beggs, A.H.; Moyer, M.; Scherpf, S.; Heindrich, K.; Bettecken, T.; Meng, G.; Müller, C.R.; Lindlöf, M.; Kaariainen, H.; et al. The molecular basis for Duchenne versus Becker muscular dystrophy: Correlation of severity with type of deletion. Am. J. Hum. Genet. 1989, 45, 498–506. [Google Scholar]
- Le Rumeur, E. Dystrophin and the two related genetic diseases, Duchenne and Becker muscular dystrophies. Bosn. J. Basic Med. Sci. 2015, 15, 14–20. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Yang, X.; Lin, G.; Han, Y.; Li, J. Molecular genetic testing and diagnosis strategies for dystrophinopathies in the era of next generation sequencing. Clin. Chim. Acta 2019, 491, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Heald, A.; Anderson, L.V.; Bushby, K.M.; Shaw, P.J. Becker muscular dystrophy with onset after 60 years. Neurology 1994, 44, 2388–2390. [Google Scholar] [CrossRef] [PubMed]
- Mias-Lucquin, D.; Dos Santos Morais, R.; Chéron, A.; Lagarrigue, M.; Winder, S.J.; Chenuel, T.; Pérez, J.; Appavou, M.S.; Martel, A.; Alviset, G.; et al. How the central domain of dystrophin acts to bridge F-actin to sarcolemmal lipids. J. Struct. Biol. 2020, 209, 107411. [Google Scholar] [CrossRef] [PubMed]
- Kyrychenko, V.; Kyrychenko, S.; Tiburcy, M.; Shelton, J.M.; Long, C.; Schneider, J.W.; Zimmermann, W.H.; Bassel-Duby, R.; Olson, E.N. Functional correction of dystrophin actin binding domain mutations by genome editing. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; Zhong, X.; Liu, L.; Cui, S.; Yang, Y.; Kong, L. Genetic analysis of 1051 Chinese families with Duchenne/Becker Muscular Dystrophy. BMC Med. Genet. 2019, 20, 139. [Google Scholar] [CrossRef] [Green Version]
- Vieitez, I.; Gallano, P.; Gonzalez-Quereda, L.; Borrego, S.; Marcos, I.; Millan, J.M.; Jairo, T.; Prior, C.; Molano, J.; Trujillo-Tiebas, M.J.; et al. Mutational spectrum of Duchenne muscular dystrophy in Spain: Study of 284 cases. Neurologia 2017, 32, 377–385. [Google Scholar] [CrossRef]
- Neri, M.; Rossi, R.; Trabanelli, C.; Mauro, A.; Selvatici, R.; Falzarano, M.S.; Spedicato, N.; Margutti, A.; Rimessi, P.; Fortunato, F.; et al. The Genetic Landscape of Dystrophin Mutations in Italy: A Nationwide Study. Front. Genet. 2020, 11, 131. [Google Scholar] [CrossRef]
- Aartsma-Rus, A.; Ginjaar, I.B.; Bushby, K. The importance of genetic diagnosis for Duchenne muscular dystrophy. J. Med. Genet. 2016, 53, 145. [Google Scholar] [CrossRef]
- Lalic, T.; Vossen, R.H.A.M.; Coffa, J.; Schouten, J.P.; Guc-Scekic, M.; Radivojevic, D.; Djurisic, M.; Breuning, M.H.; White, S.J.; den Dunnen, J.T. Deletion and duplication screening in the DMD gene using MLPA. Eur. J. Hum. Genet. 2005, 13, 1231–1234. [Google Scholar] [CrossRef]
- Varga, R.-E.; Mumtaz, R.; Jahic, A.; Rudenskaya, G.E.; Sánchez-Ferrero, E.; Auer-Grumbach, M.; Hübner, C.A.; Beetz, C. MLPA-based evidence for sequence gain: Pitfalls in confirmation and necessity for exclusion of false positives. Anal. Biochem. 2012, 421, 799–801. [Google Scholar] [CrossRef] [PubMed]
- Bovolenta, M.; Neri, M.; Fini, S.; Fabris, M.; Trabanelli, C.; Venturoli, A.; Martoni, E.; Bassi, E.; Spitali, P.; Brioschi, S.; et al. A novel custom high density-comparative genomic hybridization array detects common rearrangements as well as deep intronic mutations in dystrophinopathies. BMC Genom. 2008, 9, 572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanveer, N.; Sharma, M.C.; Sarkar, C.; Gulati, S.; Kalra, V.; Singh, S.; Bhatia, R. Diagnostic utility of skin biopsy in dystrophinopathies. Clin. Neurol. Neurosurg. 2009, 111, 496–502. [Google Scholar] [CrossRef] [PubMed]
- Iskandar, K.; Dwianingsih, E.K.; Pratiwi, L.; Kalim, A.S.; Mardhiah, H.; Putranti, A.H.; Nurputra, D.K.; Triono, A.; Herini, E.S.; Malueka, R.G.; et al. The analysis of DMD gene deletions by multiplex PCR in Indonesian DMD/BMD patients: The era of personalized medicine. BMC Res. Notes 2019, 12, 704. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Wang, H.; Xiao, B.; Wei, W.; Liu, Y.; Ye, H.; Ying, X.; Chen, Y.; Liu, X.; Ji, X.; et al. Novel noncontiguous duplications identified with a comprehensive mutation analysis in the DMD gene by DMD gene-targeted sequencing. Gene 2018, 645, 113–118. [Google Scholar] [CrossRef]
- Ginsberg, M.R.; McCarty, A.J.; Lacomis, D.; Abdel-Hamid, H.Z. Duchenne muscular dystrophy caused by a novel deep intronic DMD mutation. Muscle Nerve 2018, 57, e136–e138. [Google Scholar] [CrossRef]
- Dumont, N.A.; Wang, Y.X.; von Maltzahn, J.; Pasut, A.; Bentzinger, C.F.; Brun, C.E.; Rudnicki, M.A. Dystrophin expression in muscle stem cells regulates their polarity and asymmetric division. Nat. Med. 2015, 21, 1455–1463. [Google Scholar] [CrossRef] [Green Version]
- Chang, N.; Sincennes, M.-C.; Chevalier, F.; Brun, C.; Lacaria, M.; Segalés, J.; Muñoz-Cánovez, P.; Ming, H.; Rudnicki, M. The Dystrophin Glycoprotein Complex Regulates the Epigenetic Activation of Muscle Stem Cell Commitment. Cell Stem Cell 2018, 22, 755–768. [Google Scholar] [CrossRef] [Green Version]
- Chang, N.C.; Chevalier, F.P.; Rudnicki, M.A. Satellite Cells in Muscular Dystrophy—Lost in Polarity. Trends Mol. Med. 2016, 22, 479–496. [Google Scholar] [CrossRef] [Green Version]
- Mu, X.; Tang, Y.; Lu, A.; Takayama, K.; Usas, A.; Wang, B.; Weiss, K.; Huard, J. The role of Notch signaling in muscle progenitor cell depletion and the rapid onset of histopathology in muscular dystrophy. Hum. Mol. Genet. 2015, 24, 2923–2937. [Google Scholar] [CrossRef] [Green Version]
- Biressi, S.; Miyabara, E.H.; Gopinath, S.D.; Carlig, P.M.; Rando, T.A. A Wnt-TGFβ2 axis induces a fibrogenic program in muscle stem cells from dystrophic mice. Sci. Transl. Med. 2014, 6, 267ra176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almada, A.E.; Wagers, A.J. Molecular circuitry of stem cell fate in skeletal muscle regeneration, ageing and disease. Nat. Rev. Mol. Cell Biol. 2016, 17, 267–279. [Google Scholar] [CrossRef] [Green Version]
- Dogra, C.; Changotra, H.; Wergedal, J.E.; Kumar, A. Regulation of phosphatidylinositol 3-kinase (PI3K)/Akt and nuclear factor-kappa B signaling pathways in dystrophin-deficient skeletal muscle in response to mechanical stretch. J. Cell. Physiol. 2006, 208, 575–585. [Google Scholar] [CrossRef]
- Kumar, A.; Boriek, A.M. Mechanical stress activates the nuclear factor-kappaB pathway in skeletal muscle fibers: A possible role in Duchenne muscular dystrophy. FASEB J. 2003, 17, 386–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Malhotra, S.; Kumar, A. Nuclear factor-kappa B signaling in skeletal muscle atrophy. J. Mol. Med. 2008, 86, 1113–1126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, A.; Fueki, N.; Shiba, N.; Motoki, H.; Miyazaki, D.; Nishizawa, H.; Echigoya, Y.; Yokota, T.; Aoki, Y.; Takeda, S.i. Deletion of exons 3–9 encompassing a mutational hot spot in the DMD gene presents an asymptomatic phenotype, indicating a target region for multiexon skipping therapy. J. Hum. Genet. 2016, 61, 663–667. [Google Scholar] [CrossRef] [PubMed]
- Madaro, L.; Torcinaro, A.; De Bardi, M.; Contino, F.F.; Pelizzola, M.; Diaferia, G.R.; Imeneo, G.; Bouche, M.; Puri, P.L.; De Santa, F. Macrophages fine tune satellite cell fate in dystrophic skeletal muscle of mdx mice. PLoS Genet. 2019, 15, e1008408. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Tsai, S.Y.; Tsai, M.J. COUP-TFII regulates satellite cell function and muscular dystrophy. J. Clin. Investig. 2016, 126, 3929–3941. [Google Scholar] [CrossRef] [Green Version]
- Petrany, M.J.; Song, T.; Sadayappan, S.; Millay, D.P. Myocyte-derived Myomaker expression is required for regenerative fusion but exacerbates membrane instability in dystrophic myofibers. JCI Insight 2020, 5. [Google Scholar] [CrossRef] [Green Version]
- Lu, A.; Poddar, M.; Tang, Y.; Proto, J.D.; Sohn, J.; Mu, X.; Oyster, N.; Wang, B.; Huard, J. Rapid depletion of muscle progenitor cells in dystrophic mdx/utrophin-/- mice. Hum. Mol. Genet. 2014, 23, 4786–4800. [Google Scholar] [CrossRef] [Green Version]
- Rodgers, B.D.; Bishaw, Y.; Kagel, D.; Ramos, J.N.; Maricelli, J.W. Micro-dystrophin Gene Therapy Partially Enhances Exercise Capacity in Older Adult mdx Mice. Mol. Ther. Methods Clin. Dev. 2020, 17, 122–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pini, V.; Morgan, J.E.; Muntoni, F.; O’Neill, H.C. Genome Editing and Muscle Stem Cells as a Therapeutic Tool for Muscular Dystrophies. Curr. Stem Cell Rep. 2017, 3, 137–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, C.; Serra, C.; Lee, G.; Wagner, K.R. Stem cell-based therapies for Duchenne muscular dystrophy. Exp. Neurol. 2020, 323, 113086. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.P.; Youngblood, D.S.; Hassinger, J.N.; Lovejoy, C.E.; Nelson, M.H.; Iversen, P.L.; Moulton, H.M. Cell-penetrating peptides as transporters for morpholino oligomers: Effects of amino acid composition on intracellular delivery and cytotoxicity. Nucleic Acids Res. 2007, 35, 5182–5191. [Google Scholar] [CrossRef] [Green Version]
- Malik, V.; Rodino-Klapac, L.R.; Viollet, L.; Wall, C.; King, W.; Al-Dahhak, R.; Lewis, S.; Shilling, C.J.; Kota, J.; Serrano-Munuera, C.; et al. Gentamicin-induced readthrough of stop codons in duchenne muscular dystrophy. Ann. Neurol. 2010, 67, 771–780. [Google Scholar] [CrossRef] [PubMed]
- Barton-Davis, E.R.; Cordier, L.; Shoturma, D.I.; Leland, S.E.; Sweeney, H.L. Aminoglycoside antibiotics restore dystrophin function to skeletal muscles of mdx mice. J. Clin. Investig. 1999, 104, 375–381. [Google Scholar] [CrossRef] [Green Version]
- Wagner, K.R.; Hamed, S.; Hadley, D.W.; Gropman, A.L.; Burstein, A.H.; Escolar, D.M.; Hoffman, E.P.; Fischbeck, K.H. Gentamicin treatment of Duchenne and Becker muscular dystrophy due to nonsense mutations. Ann. Neurol. 2001, 49, 706–711. [Google Scholar] [CrossRef]
- Grages, S.M.; Bell, M.; Berlau, D.J. New and emerging pharmacotherapy for duchenne muscular dystrophy: A focus on synthetic therapeutics. Expert Opin. Pharmacother. 2020, 21, 841–851. [Google Scholar] [CrossRef]
- Shimizu-Motohashi, Y.; Miyatake, S.; Komaki, H.; Takeda, S.i.; Aoki, Y. Recent advances in innovative therapeutic approaches for Duchenne muscular dystrophy: From discovery to clinical trials. Am. J. Transl. Res. 2016, 8, 2471–2489. [Google Scholar]
- Taguchi, A.; Hamada, K.; Shiozuka, M.; Kobayashi, M.; Murakami, S.; Takayama, K.; Taniguchi, A.; Usui, T.; Matsuda, R.; Hayashi, Y. Structure-Activity Relationship Study of Leucyl-3-epi-deoxynegamycin for Potent Premature Termination Codon Readthrough. ACS Med. Chem. Lett. 2017, 8, 1060–1065. [Google Scholar] [CrossRef]
- Hamada, K.; Omura, N.; Taguchi, A.; Baradaran-Heravi, A.; Kotake, M.; Arai, M.; Takayama, K.; Taniguchi, A.; Roberge, M.; Hayashi, Y. New Negamycin-Based Potent Readthrough Derivative Effective against TGA-Type Nonsense Mutations. ACS Med. Chem. Lett. 2019, 10, 1450–1456. [Google Scholar] [CrossRef] [PubMed]
- Mercuri, E.; Muntoni, F.; Osorio, A.N.; Tulinius, M.; Buccella, F.; Morgenroth, L.P.; Gordish-Dressman, H.; Jiang, J.; Trifillis, P.; Zhu, J.; et al. Safety and effectiveness of ataluren: Comparison of results from the STRIDE Registry and CINRG DMD Natural History Study. J. Comp. Eff. Res. 2020, 9, 341–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aartsma-Rus, A.; Straub, V.; Hemmings, R.; Haas, M.; Schlosser-Weber, G.; Stoyanova-Beninska, V.; Mercuri, E.; Muntoni, F.; Sepodes, B.; Vroom, E.; et al. Development of Exon Skipping Therapies for Duchenne Muscular Dystrophy: A Critical Review and a Perspective on the Outstanding Issues. Nucleic Acid Ther. 2017, 27, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Asher, D.R.; Thapa, K.; Dharia, S.D.; Khan, N.; Potter, R.A.; Rodino-Klapac, L.R.; Mendell, J.R. Clinical development on the frontier: Gene therapy for duchenne muscular dystrophy. Expert Opin. Biol. Ther. 2020, 20, 263–274. [Google Scholar] [CrossRef]
- Muntoni, F.; Wood, M.J.A. Targeting RNA to treat neuromuscular disease. Nat. Rev. Drug Discov. 2011, 10, 621–637. [Google Scholar] [CrossRef]
- Shimizu-Motohashi, Y.; Komaki, H.; Motohashi, N.; Takeda, S.I.; Yokota, T.; Aoki, Y. Restoring Dystrophin Expression in Duchenne Muscular Dystrophy: Current Status of Therapeutic Approaches. J. Pers. Med. 2019, 9, 1. [Google Scholar] [CrossRef] [Green Version]
- Moulton, H.M.; Moulton, J.D. Morpholinos and their peptide conjugates: Therapeutic promise and challenge for Duchenne muscular dystrophy. Biochim. Biophys. Acta 2010, 1798, 2296–2303. [Google Scholar] [CrossRef] [Green Version]
- Lu, Q.-L.; Yokota, T.; Takeda, S.I.; Garcia, L.; Muntoni, F.; Partridge, T. The status of exon skipping as a therapeutic approach to duchenne muscular dystrophy. Mol. Ther. J. Am. Soc. Gene Ther. 2011, 19, 9–15. [Google Scholar] [CrossRef]
- Yokota, T.; Lu, Q.-L.; Partridge, T.; Kobayashi, M.; Nakamura, A.; Takeda, S.; Hoffman, E. Efficacy of systemic morpholino exon-skipping in duchenne dystrophy dogs. Ann. Neurol. 2009, 65, 667–676. [Google Scholar] [CrossRef]
- Aoki, Y.; Nakamura, A.; Yokota, T.; Saito, T.; Okazawa, H.; Nagata, T.; Takeda, S.I. In-frame Dystrophin Following Exon 51-Skipping Improves Muscle Pathology and Function in the Exon 52–Deficient mdx Mouse. Mol. Ther. 2010, 18, 1995–2005. [Google Scholar] [CrossRef]
- Aartsma-Rus, A.; Krieg, A.M. FDA Approves Eteplirsen for Duchenne Muscular Dystrophy: The Next Chapter in the Eteplirsen Saga. Nucleic Acid Ther. 2017, 27, 1–3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendell, J.R.; Goemans, N.; Lowes, L.P.; Alfano, L.N.; Berry, K.; Shao, J.; Kaye, E.M.; Mercuri, E. Longitudinal effect of eteplirsen versus historical control on ambulation in Duchenne muscular dystrophy. Ann. Neurol. 2016, 79, 257–271. [Google Scholar] [CrossRef] [PubMed]
- Heo, Y.-A. Golodirsen: First Approval. Drugs 2020, 80, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Frank, D.E.; Schnell, F.J.; Akana, C.; El-Husayni, S.H.; Desjardins, C.A.; Morgan, J.; Charleston, J.S.; Sardone, V.; Domingos, J.; Dickson, G.; et al. Increased dystrophin production with golodirsen in patients with Duchenne muscular dystrophy. Neurology 2020, 94, e2270–e2282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aartsma-Rus, A.; Corey, D.R. The 10th Oligonucleotide Therapy Approved: Golodirsen for Duchenne Muscular Dystrophy. Nucleic Acid Ther. 2020, 30, 67–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roshmi, R.R.; Yokota, T. Viltolarsen for the treatment of Duchenne muscular dystrophy. Drugs Today 2019, 55, 627–639. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, S. Viltolarsen: First Approval. Drugs 2020, 80, 1027–1031. [Google Scholar] [CrossRef]
- McDonald, C.M.; Wong, B.; Flanigan, K.M.; Wilson, R.; de Kimpe, S.; Lourbakos, A.; Lin, Z.; Campion, G.; DEMAND V study group. Placebo-controlled Phase 2 Trial of Drisapersen for Duchenne Muscular Dystrophy. Ann. Clin. Transl. Neurol. 2018, 5, 913–926. [Google Scholar] [CrossRef] [Green Version]
- Bosgra, S.; Sipkens, J.; de Kimpe, S.; den Besten, C.; Datson, N.; van Deutekom, J. The Pharmacokinetics of 2′-O-Methyl Phosphorothioate Antisense Oligonucleotides: Experiences from Developing Exon Skipping Therapies for Duchenne Muscular Dystrophy. Nucleic Acid Ther. 2019, 29, 305–322. [Google Scholar] [CrossRef]
- Amantana, A.; Moulton, H.M.; Cate, M.L.; Reddy, M.T.; Whitehead, T.; Hassinger, J.N.; Youngblood, D.S.; Iversen, P.L. Pharmacokinetics, Biodistribution, Stability and Toxicity of a Cell-Penetrating Peptide−Morpholino Oligomer Conjugate. Bioconjug. Chem. 2007, 18, 1325–1331. [Google Scholar] [CrossRef]
- Gait, M.J.; Arzumanov, A.A.; McClorey, G.; Godfrey, C.; Betts, C.; Hammond, S.; Wood, M.J.A. Cell-Penetrating Peptide Conjugates of Steric Blocking Oligonucleotides as Therapeutics for Neuromuscular Diseases from a Historical Perspective to Current Prospects of Treatment. Nucleic Acid Ther. 2018, 29, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsoumpra, M.K.; Fukumoto, S.; Matsumoto, T.; Takeda, S.; Wood, M.J.A.; Aoki, Y. Peptide-conjugate antisense based splice-correction for Duchenne muscular dystrophy and other neuromuscular diseases. EBioMedicine 2019, 45, 630–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaw, K.; Greer, K.; Aung-Htut, M.T.; Mitrpant, C.; Veedu, R.N.; Fletcher, S.; Wilton, S.D. Consequences of Making the Inactive Active Through Changes in Antisense Oligonucleotide Chemistries. Front. Genet. 2019, 10, 1249. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Han, G.; Ning, H.; Song, J.; Ran, N.; Yi, X.; Seow, Y.; Yin, H. Glycine Enhances Satellite Cell Proliferation, Cell Transplantation, and Oligonucleotide Efficacy in Dystrophic Muscle. Mol. Ther. 2020, 28, 1339–1358. [Google Scholar] [CrossRef]
- Lu, Q.L.; Rabinowitz, A.; Chen, Y.C.; Yokota, T.; Yin, H.; Alter, J.; Jadoon, A.; Bou-Gharios, G.; Partridge, T. Systemic delivery of antisense oligoribonucleotide restores dystrophin expression in body-wide skeletal muscles. Proc. Natl. Acad. Sci. USA 2005, 102, 198–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, Q.; Yokota, T. Antisense oligonucleotides for the treatment of cardiomyopathy in Duchenne muscular dystrophy. Am. J. Transl. Res. 2019, 11, 1202–1218. [Google Scholar]
- Yin, H.; Moulton, H.M.; Seow, Y.; Boyd, C.; Boutilier, J.; Iverson, P.; Wood, M.J.A. Cell-penetrating peptide-conjugated antisense oligonucleotides restore systemic muscle and cardiac dystrophin expression and function. Hum. Mol. Genet. 2008, 17, 3909–3918. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Xu, M.; Cui, Y.; Huang, C.; Sun, M. Arginine-rich membrane-permeable peptides are seriously toxic. Pharmacol. Res. Perspect. 2017, 5. [Google Scholar] [CrossRef]
- Duan, D. Systemic AAV Micro-dystrophin Gene Therapy for Duchenne Muscular Dystrophy. Mol. Ther. 2018, 26, 2337–2356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tedesco, F.S. Human artificial chromosomes for Duchenne muscular dystrophy and beyond: Challenges and hopes. Chromosome Res. 2015, 23, 135–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harper, S.Q.; Hauser, M.A.; DelloRusso, C.; Duan, D.; Crawford, R.W.; Phelps, S.F.; Harper, H.A.; Robinson, A.S.; Engelhardt, J.F.; Brooks, S.V.; et al. Modular flexibility of dystrophin: Implications for gene therapy of Duchenne muscular dystrophy. Nat. Med. 2002, 8, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Hermonat, P.L.; Muzyczka, N. Use of adeno-associated virus as a mammalian DNA cloning vector: Transduction of neomycin resistance into mammalian tissue culture cells. Proc. Natl. Acad. Sci. USA 1984, 81, 6466–6470. [Google Scholar] [CrossRef] [Green Version]
- Le Guiner, C.; Servais, L.; Montus, M.; Larcher, T.; Fraysse, B.; Moullec, S.; Allais, M.; François, V.; Dutilleul, M.; Malerba, A.; et al. Long-term microdystrophin gene therapy is effective in a canine model of Duchenne muscular dystrophy. Nat. Commun. 2017, 8, 16105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- England, S.B.; Nicholson, L.V.; Johnson, M.A.; Forrest, S.M.; Love, D.R.; Zubrzycka-Gaarn, E.E.; Bulman, D.E.; Harris, J.B.; Davies, K.E. Very mild muscular dystrophy associated with the deletion of 46% of dystrophin. Nature 1990, 343, 180–182. [Google Scholar] [CrossRef] [PubMed]
- Koo, T.; Popplewell, L.; Athanasopoulos, T.; Dickson, G. Triple trans-splicing adeno-associated virus vectors capable of transferring the coding sequence for full-length dystrophin protein into dystrophic mice. Hum. Gene Ther. 2014, 25, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Kornegay, J.N.; Li, J.; Bogan, J.R.; Bogan, D.J.; Chen, C.; Zheng, H.; Wang, B.; Qiao, C.; Howard, J.F.; Xiao, X. Widespread muscle expression of an AAV9 human mini-dystrophin vector after intravenous injection in neonatal dystrophin-deficient dogs. Mol. Ther. J. Am. Soc. Gene Ther. 2010, 18, 1501–1508. [Google Scholar] [CrossRef]
- Martino, A.T.; Suzuki, M.; Markusic, D.M.; Zolotukhin, I.; Ryals, R.C.; Moghimi, B.; Ertl, H.C.J.; Muruve, D.A.; Lee, B.; Herzog, R.W. The genome of self-complementary adeno-associated viral vectors increases Toll-like receptor 9-dependent innate immune responses in the liver. Blood 2011, 117, 6459–6468. [Google Scholar] [CrossRef] [Green Version]
- Mendell, J.R.; Campbell, K.; Rodino-Klapac, L.; Sahenk, Z.; Shilling, C.; Lewis, S.; Bowles, D.; Gray, S.; Li, C.; Galloway, G.; et al. Dystrophin immunity in Duchenne’s muscular dystrophy. N. Engl. J. Med. 2010, 363, 1429–1437. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Morales, L.; Malik, A.S.; Mead, A.F.; Greer, C.D.; Mitchell, M.A.; Petrov, M.T.; Su, L.T.; Choi, M.E.; Rosenblum, S.T.; et al. Non-immunogenic utrophin gene therapy for the treatment of muscular dystrophy animal models. Nat. Med. 2019, 25, 1505–1511. [Google Scholar] [CrossRef]
- Bowles, D.E.; McPhee, S.W.; Li, C.; Gray, S.J.; Samulski, J.J.; Camp, A.S.; Li, J.; Wang, B.; Monahan, P.E.; Rabinowitz, J.E.; et al. Phase 1 gene therapy for Duchenne muscular dystrophy using a translational optimized AAV vector. Mol. Ther. 2012, 20, 443–455. [Google Scholar] [CrossRef] [Green Version]
- Chandran, J.S.; Sharp, P.S.; Karyka, E.; Aves-Cruzeiro, J.; Coldicott, I.; Castelli, L.; Hautbergue, G.; Collins, M.O.; Azzouz, M. Site Specific Modification of Adeno-Associated Virus Enables Both Fluorescent Imaging of Viral Particles and Characterization of the Capsid Interactome. Sci. Rep. 2017, 7, 14766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooney, A.L.; Thornell, I.M.; Singh, B.K.; Shah, V.S.; Stoltz, D.A.; McCray, P.B., Jr.; Zabner, J.; Sinn, P.L. A Novel AAV-mediated Gene Delivery System Corrects CFTR Function in Pigs. Am. J. Respir. Cell Mol. Biol. 2019, 61, 747–754. [Google Scholar] [CrossRef] [PubMed]
- Kazuki, Y.; Hoshiya, H.; Takiguchi, M.; Abe, S.; Iida, Y.; Osaki, M.; Katoh, M.; Hiratsuka, M.; Shirayoshi, Y.; Hiramatsu, K.; et al. Refined human artificial chromosome vectors for gene therapy and animal transgenesis. Gene Ther. 2011, 18, 384–393. [Google Scholar] [CrossRef] [PubMed]
- Sinenko, S.A.; Ponomartsev, S.V.; Tomilin, A.N. Human artificial chromosomes for pluripotent stem cell-based tissue replacement therapy. Exp. Cell Res. 2020, 389, 111882. [Google Scholar] [CrossRef] [PubMed]
- Babačić, H.; Mehta, A.; Merkel, O.; Schoser, B. CRISPR-cas gene-editing as plausible treatment of neuromuscular and nucleotide-repeat-expansion diseases: A systematic review. PLoS ONE 2019, 14, e0212198. [Google Scholar] [CrossRef] [Green Version]
- Ran, F.A.; Cong, L.; Yan, W.X.; Scott, D.A.; Gootenberg, J.S.; Kriz, A.J.; Zetsche, B.; Shalem, O.; Wu, X.; Makarova, K.S.; et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature 2015, 520, 186–191. [Google Scholar] [CrossRef]
- Xie, H.; Tang, L.; He, X.; Liu, X.; Zhou, C.; Liu, J.; Ge, X.; Li, J.; Liu, C.; Zhao, J.; et al. SaCas9 Requires 5′-NNGRRT-3′ PAM for Sufficient Cleavage and Possesses Higher Cleavage Activity than SpCas9 or FnCpf1 in Human Cells. Biotechnol. J. 2018, 13, e1700561. [Google Scholar] [CrossRef]
- Young, C.S.; Hicks, M.R.; Ermolova, N.V.; Nakano, H.; Jan, M.; Younesi, S.; Karumbayaram, S.; Kumagai-Cresse, C.; Wang, D.; Zack, J.A.; et al. A Single CRISPR-Cas9 Deletion Strategy that Targets the Majority of DMD Patients Restores Dystrophin Function in hiPSC-Derived Muscle Cells. Cell Stem Cell 2016, 18, 533–540. [Google Scholar] [CrossRef] [Green Version]
- Min, Y.L.; Li, H.; Rodriguez-Caycedo, C.; Mireault, A.A.; Huang, J.; Shelton, J.M.; McAnally, J.R.; Amoasii, L.; Mammen, P.P.A.; Bassel-Duby, R.; et al. CRISPR-Cas9 corrects Duchenne muscular dystrophy exon 44 deletion mutations in mice and human cells. Sci. Adv. 2019, 5, eaav4324. [Google Scholar] [CrossRef] [Green Version]
- Zhu, P.; Wu, F.; Mosenson, J.; Zhang, H.; He, T.C.; Wu, W.S. CRISPR/Cas9-Mediated Genome Editing Corrects Dystrophin Mutation in Skeletal Muscle Stem Cells in a Mouse Model of Muscle Dystrophy. Mol. Ther. Nucleic Acids 2017, 7, 31–41. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Park, K.H.; Zhao, L.; Xu, J.; El Refaey, M.; Gao, Y.; Zhu, H.; Ma, J.; Han, R. CRISPR-mediated Genome Editing Restores Dystrophin Expression and Function in mdx Mice. Mol. Ther. 2016, 24, 564–569. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.; Conboy, M.; Park, H.M.; Jiang, F.; Kim, H.J.; Dewitt, M.A.; Mackley, V.A.; Chang, K.; Rao, A.; Skinner, C.; et al. Nanoparticle delivery of Cas9 ribonucleoprotein and donor DNA in vivo induces homology-directed DNA repair. Nat. Biomed. Eng. 2017, 1, 889–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabebordbar, M.; Zhu, K.; Cheng, J.K.W.; Chew, W.L.; Widrick, J.J.; Yan, W.X.; Maesner, C.; Wu, E.Y.; Xiao, R.; Ran, F.A.; et al. In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science 2016, 351, 407–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, C.; Liu, Y.; Ma, T.; Liu, K.; Xu, S.; Zhang, Y.; Liu, H.; La Russa, M.; Xie, M.; Ding, S.; et al. Small molecules enhance CRISPR genome editing in pluripotent stem cells. Cell Stem Cell 2015, 16, 142–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Y.; Sander, J.D.; Reyon, D.; Cascio, V.M.; Joung, J.K. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat. Biotechnol. 2014, 32, 279–284. [Google Scholar] [CrossRef] [Green Version]
- Cho, S.W.; Kim, S.; Kim, Y.; Kweon, J.; Kim, H.S.; Bae, S.; Kim, J.-S. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 2014, 24, 132–141. [Google Scholar] [CrossRef] [Green Version]
- Müller, M.; Lee, C.M.; Gasiunas, G.; Davis, T.H.; Cradick, T.J.; Siksnys, V.; Bao, G.; Cathomen, T.; Mussolino, C. Streptococcus thermophilus CRISPR-Cas9 Systems Enable Specific Editing of the Human Genome. Mol. Ther. J. Am. Soc. Gene Ther. 2016, 24, 636–644. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.M.; Cradick, T.J.; Bao, G. The Neisseria meningitidis CRISPR-Cas9 System Enables Specific Genome Editing in Mammalian Cells. Mol. Ther. J. Am. Soc. Gene Ther. 2016, 24, 645–654. [Google Scholar] [CrossRef] [Green Version]
- Terao, M.; Tamano, M.; Hara, S.; Kato, T.; Kinoshita, M.; Takada, S. Utilization of the CRISPR/Cas9 system for the efficient production of mutant mice using crRNA/tracrRNA with Cas9 nickase and FokI-dCas9. Exp. Anim. 2016, 65, 275–283. [Google Scholar] [CrossRef] [Green Version]
- Watt, D.J.; Morgan, J.E.; Partridge, T.A. Use of mononuclear precursor cells to insert allogeneic genes into growing mouse muscles. Muscle Nerve 1984, 7, 741–750. [Google Scholar] [CrossRef]
- Partridge, T.A.; Morgan, J.E.; Coulton, G.R.; Hoffman, E.P.; Kunkel, L.M. Conversion of mdx myofibres from dystrophin-negative to -positive by injection of normal myoblasts. Nature 1989, 337, 176–179. [Google Scholar] [CrossRef]
- Karpati, G.; Ajdukovic, D.; Arnold, D.; Gledhill, R.B.; Guttmann, R.; Holland, P.; Koch, P.A.; Shoubridge, E.; Spence, D.; Vanasse, M.; et al. Myoblast transfer in Duchenne muscular dystrophy. Ann. Neurol. 1993, 34, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Skuk, D.; Tremblay, J.P. Myoblast transplantation: The current status of a potential therapeutic tool for myopathies. J. Muscle Res. Cell Motil. 2003, 24, 285–300. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.H.; Mouly, V.; Cooper, R.N.; Mamchaoui, K.; Bigot, A.; Shay, J.W.; Di Santo, J.P.; Butler-Browne, G.S.; Wright, W.E. Cellular senescence in human myoblasts is overcome by human telomerase reverse transcriptase and cyclin-dependent kinase 4: Consequences in aging muscle and therapeutic strategies for muscular dystrophies. Aging Cell 2007, 6, 515–523. [Google Scholar] [CrossRef]
- Sacco, A.; Doyonnas, R.; Kraft, P.; Vitorovic, S.; Blau, H.M. Self-renewal and expansion of single transplanted muscle stem cells. Nature 2008, 456, 502–506. [Google Scholar] [CrossRef] [Green Version]
- Elster, J.L.; Rathbone, C.R.; Liu, Z.; Liu, X.; Barrett, H.H.; Rhoads, R.P.; Allen, R.E. Skeletal muscle satellite cell migration to injured tissue measured with 111In-oxine and high-resolution SPECT imaging. J. Muscle Res. Cell Motil. 2013, 34, 417–427. [Google Scholar] [CrossRef] [Green Version]
- Morgan, J.; Rouche, A.; Bausero, P.; Houssaïni, A.; Gross, J.; Fiszman, M.Y.; Alameddine, H.S. MMP-9 overexpression improves myogenic cell migration and engraftment. Muscle Nerve 2010, 42, 584–595. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, P.M.; Havenstrite, K.L.; Magnusson, K.E.; Sacco, A.; Leonardi, N.A.; Kraft, P.; Nguyen, N.K.; Thrun, S.; Lutolf, M.P.; Blau, H.M. Substrate elasticity regulates skeletal muscle stem cell self-renewal in culture. Science 2010, 329, 1078–1081. [Google Scholar] [CrossRef] [Green Version]
- Lutolf, M.P.; Gilbert, P.M.; Blau, H.M. Designing materials to direct stem-cell fate. Nature 2009, 462, 433–441. [Google Scholar] [CrossRef] [Green Version]
- Ricotti, L.; Polini, A.; Genchi, G.G.; Ciofani, G.; Iandolo, D.; Vazão, H.; Mattoli, V.; Ferreira, L.; Menciassi, A.; Pisignano, D. Proliferation and skeletal myotube formation capability of C2C12 and H9c2 cells on isotropic and anisotropic electrospun nanofibrous PHB scaffolds. Biomed. Mater. 2012, 7, 035010. [Google Scholar] [CrossRef] [PubMed]
- Ku, S.H.; Lee, S.H.; Park, C.B. Synergic effects of nanofiber alignment and electroactivity on myoblast differentiation. Biomaterials 2012, 33, 6098–6104. [Google Scholar] [CrossRef] [PubMed]
- Leung, M.; Cooper, A.; Jana, S.; Tsao, C.T.; Petrie, T.A.; Zhang, M. Nanofiber-based in vitro system for high myogenic differentiation of human embryonic stem cells. Biomacromolecules 2013, 14, 4207–4216. [Google Scholar] [CrossRef]
- Chal, J.; Oginuma, M.; Al Tanoury, Z.; Gobert, B.; Sumara, O.; Hick, A.; Bousson, F.; Zidouni, Y.; Mursch, C.; Moncuquet, P.; et al. Differentiation of pluripotent stem cells to muscle fiber to model Duchenne muscular dystrophy. Nat. Biotechnol. 2015, 33, 962–969. [Google Scholar] [CrossRef] [Green Version]
- Darabi, R.; Santos, F.N.; Filareto, A.; Pan, W.; Koene, R.; Rudnicki, M.A.; Kyba, M.; Perlingeiro, R.C. Assessment of the myogenic stem cell compartment following transplantation of Pax3/Pax7-induced embryonic stem cell-derived progenitors. Stem Cells 2011, 29, 777–790. [Google Scholar] [CrossRef] [Green Version]
- Dellavalle, A.; Maroli, G.; Covarello, D.; Azzoni, E.; Innocenzi, A.; Perani, L.; Antonini, S.; Sambasivan, R.; Brunelli, S.; Tajbakhsh, S.; et al. Pericytes resident in postnatal skeletal muscle differentiate into muscle fibres and generate satellite cells. Nat. Commun. 2011, 2, 499. [Google Scholar] [CrossRef] [Green Version]
- Sharp, P.S.; Bye-a-Jee, H.; Wells, D.J. Physiological characterization of muscle strength with variable levels of dystrophin restoration in mdx mice following local antisense therapy. Mol. Ther. J. Am. Soc. Gene Ther. 2011, 19, 165–171. [Google Scholar] [CrossRef]
- Godfrey, C.; Muses, S.; McClorey, G.; Wells, K.E.; Coursindel, T.; Terry, R.L.; Betts, C.; Hammond, S.; O’Donovan, L.; Hildyard, J.; et al. How much dystrophin is enough: The physiological consequences of different levels of dystrophin in the mdx mouse. Hum. Mol. Genet. 2015, 24, 4225–4237. [Google Scholar] [CrossRef] [Green Version]
- Van Putten, M.; Hulsker, M.; Nadarajah, V.D.; van Heiningen, S.H.; van Huizen, E.; van Iterson, M.; Admiraal, P.; Messemaker, T.; den Dunnen, J.T.; AC’t Hoen, P.; et al. The effects of low levels of dystrophin on mouse muscle function and pathology. PLoS ONE 2012, 7, e31937. [Google Scholar] [CrossRef] [Green Version]
- Le Guiner, C.; Montus, M.; Servais, L.; Cherel, Y.; Francois, V.; Thibaud, J.-L.; Wary, C.; Matot, B.; Larcher, T.; Guigand, L.; et al. Forelimb treatment in a large cohort of dystrophic dogs supports delivery of a recombinant AAV for exon skipping in Duchenne patients. Mol. Ther. J. Am. Soc. Gene Ther. 2014, 22, 1923–1935. [Google Scholar] [CrossRef] [Green Version]
- Gentil, C.; Le Guiner, C.; Falcone, S.; Hogrel, J.-Y.; Peccate, C.; Lorain, S.; Benkhelifa-Ziyyat, S.; Guigand, L.; Montus, M.; Servais, L.; et al. Dystrophin Threshold Level Necessary for Normalization of Neuronal Nitric Oxide Synthase, Inducible Nitric Oxide Synthase, and Ryanodine Receptor-Calcium Release Channel Type 1 Nitrosylation in Golden Retriever Muscular Dystrophy Dystrophinopathy. Hum. Gene Ther. 2016, 27, 712–726. [Google Scholar] [CrossRef]
- Neri, M.; Torelli, S.; Brown, S.; Ugo, I.; Sabatelli, P.; Merlini, L.; Spitali, P.; Rimessi, P.; Gualandi, F.; Sewry, C.; et al. Dystrophin levels as low as 30% are sufficient to avoid muscular dystrophy in the human. Neuromuscul. Disord. NMD 2007, 17, 913–918. [Google Scholar] [CrossRef]
- Beekman, C.; Janson, A.A.; Baghat, A.; van Deutekom, J.C.; Datson, N.A. Use of capillary Western immunoassay (Wes) for quantification of dystrophin levels in skeletal muscle of healthy controls and individuals with Becker and Duchenne muscular dystrophy. PLoS ONE 2018, 13, e0195850. [Google Scholar] [CrossRef]
- Nicholson, L.V. The “rescue” of dystrophin synthesis in boys with Duchenne muscular dystrophy. Neuromuscul. Disord. NMD 1993, 3, 525–531. [Google Scholar] [CrossRef]
- Waldrop, M.A.; Gumienny, F.; El Husayni, S.; Frank, D.E.; Weiss, R.B.; Flanigan, K.M. Low-level dystrophin expression attenuating the dystrophinopathy phenotype. Neuromuscul. Disord. NMD 2018, 28, 116–121. [Google Scholar] [CrossRef]
- Wells, D.J. What is the level of dystrophin expression required for effective therapy of Duchenne muscular dystrophy? J. Muscle Res. Cell Motil. 2019, 40, 141–150. [Google Scholar] [CrossRef]
- Goldstein, J.M.; Tabebordbar, M.; Zhu, K.; Wang, L.D.; Messemer, K.A.; Peacker, B.; Ashrafi Kakhki, S.; Gonzalez-Celeiro, M.; Shwartz, Y.; Cheng, J.K.W.; et al. In Situ Modification of Tissue Stem and Progenitor Cell Genomes. Cell Rep. 2019, 27, 1254–1264. [Google Scholar] [CrossRef] [Green Version]
- Charville, G.W.; Cheung, T.H.; Yoo, B.; Santos, P.J.; Lee, G.K.; Shrager, J.B.; Rando, T.A. Ex Vivo Expansion and In Vivo Self-Renewal of Human Muscle Stem Cells. Stem Cell Rep. 2015, 5, 621–632. [Google Scholar] [CrossRef] [Green Version]
- Arnett, A.L.; Konieczny, P.; Ramos, J.N.; Hall, J.; Odom, G.; Yablonka-Reuveni, Z.; Chamberlain, J.R.; Chamberlain, J.S. Adeno-associated viral (AAV) vectors do not efficiently target muscle satellite cells. Mol. Ther. Methods Clin. Dev. 2014, 1, 14038. [Google Scholar] [CrossRef]
- Muraine, L.; Bensalah, M.; Dhiab, J.; Cordova, G.; Arandel, L.; Marhic, A.; Chapart, M.; Vasseur, S.; Benkhelifa-Ziyyat, S.; Bigot, A.; et al. Transduction Efficiency of Adeno-Associated Virus Serotypes After Local Injection in Mouse and Human Skeletal Muscle. Hum. Gene Ther. 2020, 31, 233–240. [Google Scholar] [CrossRef]
- Nance, M.E.; Shi, R.; Hakim, C.H.; Wasala, N.B.; Yue, Y.; Pan, X.; Zhang, T.; Robinson, C.A.; Duan, S.X.; Yao, G.; et al. AAV9 Edits Muscle Stem Cells in Normal and Dystrophic Adult Mice. Mol. Ther. 2019, 27, 1568–1585. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Chemical | Drug | Other Name | Sponsor | Targeted Exon | Clinical Trial Number | Trial Phase | Start Date | Completion Date |
|---|---|---|---|---|---|---|---|---|
| PMO 1 | Eteplirsen | AVI-4658, Exondys 51 | Sarepta Therapeutics | Exon 51 | NCT03218995 | 2 | August 2017 | March 2021 |
| NCT04179409 | 2 | February 2020 | September 2022 | |||||
| NCT03992430 | 3 | January 2020 | October 2024 | |||||
| NCT03985878 | 3 | June 2019 | February 2027 | |||||
| Golodirsen | SRP4053 | Sarepta Therapeutics | Exon 53 | NCT04179409 | 2 | February 2020 | September 2022 | |
| NCT02500381 | 3 | September 2016 | May 2023 | |||||
| NCT03532542 | 3 | 2 August 2018 | 10 August 2026 | |||||
| Casimersen | SRP4045 | Sarepta Therapeutics | Exon 45 | NCT04179409 | 2 | February 2020 | September 2022 | |
| NCT03532542 | 3 | 2 August 2018 | 10 August 2026 | |||||
| Viltolarsen | NCNP-01, NS-065 | Nippon Shinyaku Co Ltd. | Exon 53 | NCT03167255 | 2 | July 2017 | August 2021 | |
| NCT04060199 | 3 | April 2020 | December 2024 | |||||
| 2′-O-methyl PS | Drisapersen | PRO051, GSK2402968 | BioMarin Pharmaceutical Inc. | Exon 51 | NCT02636686 | Extension | December 2015 | January 2018 |
| DS-5141b | Daiichi Sankyo Co., Ltd. | Exon 45 | NCT02667483 | 1, 2 | October 2015 | December 2020 | ||
| PPMO 2 | SRP-5051 | Sarepta Therapeutics | Exon 51 | NCT03675126 | 1, 2 | December 2018 | July 2024 | |
| NCT04004065 | 2 | June 2019 | August 2021 | |||||
| Stereopure | Suvodirsen | WVE-210201 | Wave Life Sciences Ltd. | Exon 51 | NCT03907072 | 2, 3 | September 2019 | January 2020 |
| Sponsor | Nationwide Children’s Hospital | Solid Biosciences, LLC | Sarepta Therapeutics, Inc. | Pfizer | Kevin Flanigan | Jerry R. Mendell |
|---|---|---|---|---|---|---|
| ClinicalTrials.gov Identifier | NCT00428935 | NCT03368742 | NCT03375164 | NCT03362502 | NCT03333590 | NCT02376816 |
| Trial Title | Safety study of minidystrophin gene to treat Duchenne Muscular Dystrophy | Microdystrophin gene transfer study in adolescents and children with DMD (IGNITE DMD) | Systemic gene delivery clinical trial for Duchenne Muscular Dystrophy | A study to evaluate the safety and tolerability of PF-06939926 gene therapy in Duchenne Muscular Dystrophy | Gene transfer clinical trial to deliver rAAVrh74.MCK.GALGT2 for Duchenne Muscular Dystrophy | Clinical intramuscular gene transfer trial of rAAVrh74.MCK. microdystrophin to patients With Duchenne Muscular Dystrophy |
| Recruitment Status | Completed | Suspended (clinical hold) | Active, not recruiting | Recruiting | Active, not recruiting | Completed |
| Study Start Date | March 2006 | 6 December 2017 | 4 January 2018 | 23 January 2018 | 6 November 2017 | March 2015 |
| (Estimated) Study Completion Date | July 2010 | March 2021 | April 2021 | 26 August 2025 | November 2021 | September 2017 |
| Intervention/Treatment | Biological: rAAV2.5-CMV-minidystrophin (d3990) | Genetic: SGT-001 | Genetic: rAAVrh74.MHCK7 microdystrophin | Genetic: PF-06939926 | Biological: rAAVrh74.MCK.GALGT2 | Biological: rAAVrh74.MCK. microdystrophin |
| Enrollment | 6 participants | 16 participants | 4 participants | 15 participants | 6 participants | 2 participants |
| Patient Age | 5–12 years | 4–17 years | 3 months to 7 years | 4–12 years | 4 years and older | 7 years and older |
| Dose | Cohort 1: 2.0E10 vg/kg Cohort 2: 1.0E11 vg/kg | Ascending doses (quantitative value not reminded) | 2.0E14 vg/kg in 10 mL/kg | Ascending doses (quantitative value not reminded) | Cohort 1: 5.0E13 vg/kg Cohort 2: 1.0E14 vg/kg | Cohort 1: 3E11 vg/single foot Cohort 2: 1E12 vg/single foot |
| AAV Serotype | AAV2.5 | AAV9 | AAVrh74 | AAV9 | AAVrh74 | AAVrh74 |
| Delivery Type | Intramuscular injection into biceps muscle | Intravenous injection | Intravenous injection into peripheral arm vein | Intravenous injection | Intravascular limb infusion | Intramuscular injection into Extensor digitorum brevis (EDB) muscle |
| Primary Outcome | Safety and tolerability [88] | Safety | Safety | Safety and tolerability | Safety | Safety |
| Secondary Outcome | Minidystrophin gene expression and muscle strength test [88] | No secondary outcome yet | Microdystrophin expression and muscle motility assessment | Minidystrophin gene expression, muscle strength and quality | GALGT2 gene expression and muscle motility assessment | Transgene expression |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, C.; Shen, L.; Zhang, Z.; Xie, X. Therapeutic Strategies for Duchenne Muscular Dystrophy: An Update. Genes 2020, 11, 837. https://doi.org/10.3390/genes11080837
Sun C, Shen L, Zhang Z, Xie X. Therapeutic Strategies for Duchenne Muscular Dystrophy: An Update. Genes. 2020; 11(8):837. https://doi.org/10.3390/genes11080837
Chicago/Turabian StyleSun, Chengmei, Luoan Shen, Zheng Zhang, and Xin Xie. 2020. "Therapeutic Strategies for Duchenne Muscular Dystrophy: An Update" Genes 11, no. 8: 837. https://doi.org/10.3390/genes11080837
APA StyleSun, C., Shen, L., Zhang, Z., & Xie, X. (2020). Therapeutic Strategies for Duchenne Muscular Dystrophy: An Update. Genes, 11(8), 837. https://doi.org/10.3390/genes11080837