Biomarkers of Fabry Nephropathy: Review and Future Perspective


Abstract
:1. Introduction
2. Current Diagnostic Approaches and Follow-Up of Fabry Nephropathy
2.1. Albuminuria/Proteinuria
2.2. Serum Creatinine and Glomerular Filtration Rate (GFR)
2.3. Cystatin-C
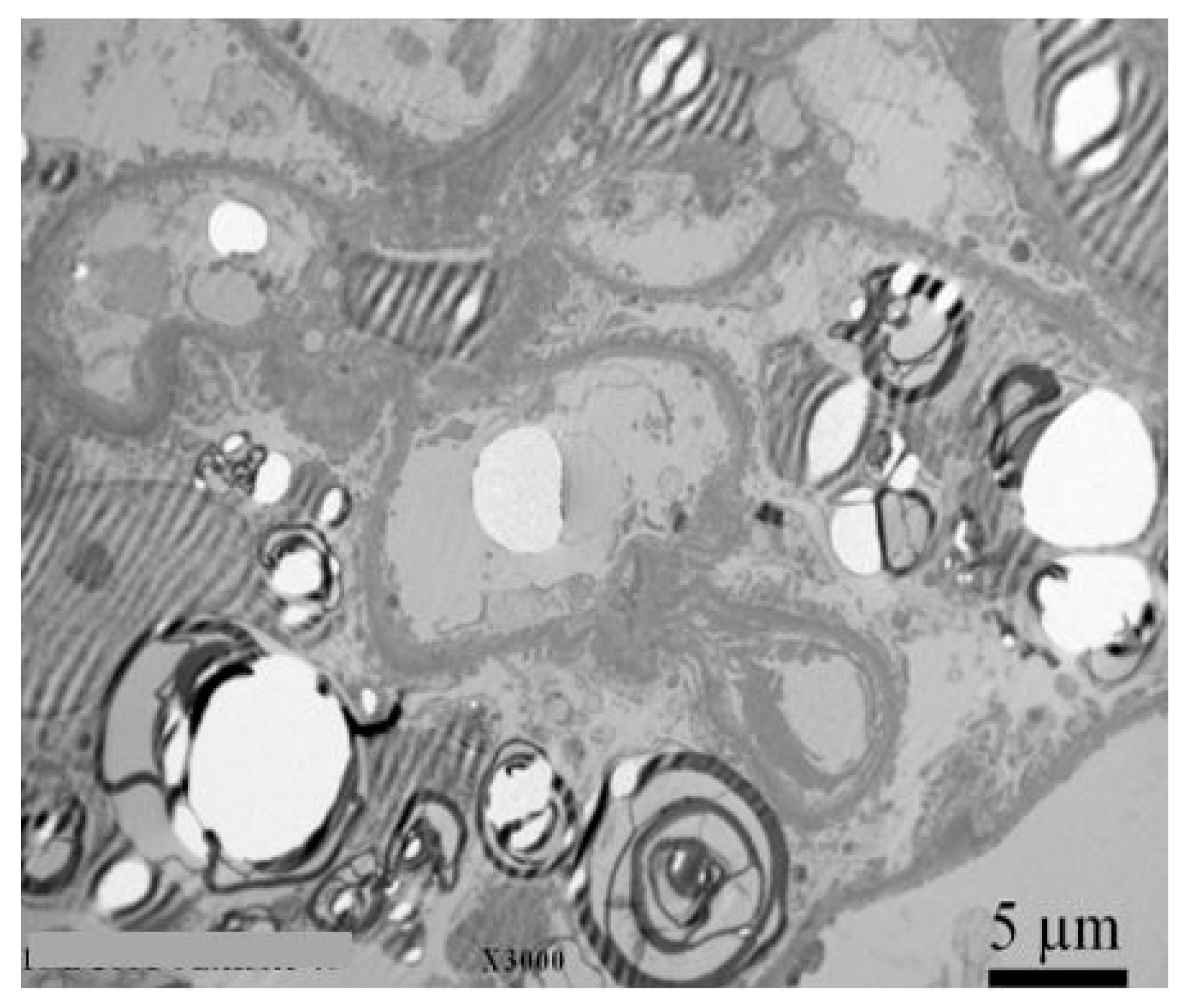
2.4. Urine Microscopy
2.5. Renal Biopsy
3. Novel Biomarkers for Predicting Development and Progression of Fabry Nephropathy
3.1. Bikunin
3.2. Tubular and Glomerular Proteins
3.3. Urine Podocytes
3.4. Promising Aproaches in Discovery of Fabry Nephropaty Biomarkers
3.4.1. Genomics
3.4.2. Transcriptomics
3.4.3. Epigenomics
3.4.4. Proteomics
3.4.5. Metabolomics
4. Discussion and Future Perspective
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| α-galactosidase A | α-Gal A |
| GLA | α-galactosidase A gene |
| ACR | albumin to creatinine ratio |
| CKD | chronic kidney disease |
| eGFR | estimated glomerular filtration rate |
| ERT | enzyme replacement therapy |
| ESRD | end stage renal disease |
| FD | Fabry disease |
| Ga2 | galabiosylceramide |
| Gb3 | globotriaosylceramide |
| lyso-Gb3 | globotriaosylsphingosine |
| SNP | single nucleotide polymorphism |
| UPod | urinary podocytes |
References
- Brady, R.O.; Gal, A.E.; Bradley, R.M.; Martensson, E.; Warshaw, A.L.; Laster, L. Enzymatic Defect in Fabry’s Disease. Ceramidetrihexosidase Deficiency. N. Engl. J. Med. 1967, 276, 1163–1167. [Google Scholar] [CrossRef] [PubMed]
- Hamers, M.N.; Westerveld, A.; Khan, M.; Tager, J.M. Characterization of α-galactosidase isoenzymes in normal and Fabry human-Chinese Hamster somatic cell hybrids. Hum. Genet. 1977, 36, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Aerts, J.M.; Groener, J.E.; Kuiper, S.; Donker-Koopman, W.E.; Strijland, A.; Ottenhoff, R.; Van Roomen, C.; Mirzaian, M.; Wijburg, F.A.; Linthorst, G.E.; et al. Elevated globotriaosylsphingosine is a hallmark of Fabry disease. Proc. Natl. Acad. Sci. USA 2008, 105, 2812–2817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rombach, S.M.; Dekker, N.; Bouwman, M.G.; Linthorst, G.E.; Zwinderman, A.H.; Wijburg, F.A.; Kuiper, S.; vd Bergh Weerman, M.A.; Groener, J.E.M.; Poorthuis, B.J.; et al. Plasma globotriaosylsphingosine: Diagnostic value and relation to clinical manifestations of Fabry disease. Biochim. Biophys. Acta 2010, 1802, 741–748. [Google Scholar] [CrossRef] [Green Version]
- Askari, H.; Kaneski, C.R.; Semino-Mora, C.; Desai, P.; Ang, A.; Kleiner, D.E.; Perlee, L.T.; Quezado, M.; Spollen, L.E.; Wustman, B.A.; et al. Cellular and tissue localization of globotriaosylceramide in Fabry disease. Virchows Arch. 2007, 451, 823–834. [Google Scholar] [CrossRef]
- Oliveira, J.P.; Ferreira, S. Multiple phenotypic domains of Fabry disease and their relevance for establishing genotype-phenotype correlations. Appl. Clin. Genet. 2019, 12, 35–50. [Google Scholar] [CrossRef] [Green Version]
- Mehta, A.; Clarke, J.T.R.; Giugliani, R.; Elliott, P.; Linhart, A.; Beck, M.; Sunder-Plassmann, G. Natural course of Fabry disease: Changing pattern of causes of death in FOS - Fabry Outcome Survey. J. Med. Genet. 2009, 46, 548–552. [Google Scholar] [CrossRef] [Green Version]
- Schiffmann, R.; Hughes, D.A.; Linthorst, G.E.; Ortiz, A.; Svarstad, E.; Warnock, D.G.; West, M.L.; Wanner, C. Screening, diagnosis, and management of patients with Fabry disease: Conclusions from a “Kidney Disease: Improving Global Outcomes” (KDIGO) Controversies Conference. Kidney Int. 2017, 91, 284–293. [Google Scholar] [CrossRef]
- Vedder, A.C.; Strijland, A.; vd Bergh Weerman, M.A.; Florquin, S.; Aerts, J.M.F.G.; Hollak, C.E.M. Manifestations of Fabry disease in placental tissue. J. Inherit. Metab. Dis. 2006, 29, 106–111. [Google Scholar] [CrossRef]
- Popli, S.; Leehey, D.J.; Molnar, Z.V.; Nawab, Z.M.; Ing, T.S. Demonstration of Fabry’s disease deposits in placenta. Am. J. Obstet. Gynecol. 1990, 162, 464–465. [Google Scholar] [CrossRef]
- Germain, D.P. Fabry disease. Orphanet J. Rare Dis. 2010, 5, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desnick, R.J. Fabry Disease: α-Galactosidase A Deficiency. In Rosenberg’s Molecular and Genetic Basis of Neurological and Psychiatric Disease: Fifth Edition; Academic Press: Cambridge, MA, USA, 2015; pp. 419–430. ISBN 9780124105294. [Google Scholar]
- Redonnet-Vernhet, I.; Ploos Van Amstel, J.K.; Jansen, R.P.M.; Wevers, R.A.; Salvayre, R.; Levade, T. Uneven X inactivation in a female monozygotic twin pair with Fabry disease and discordant expression of a novel mutation in the α-galactosidase A gene. J. Med. Genet. 1996, 33, 682–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobrovolny, R.; Dvorakova, L.; Ledvinova, J.; Magage, S.; Bultas, J.; Lubanda, J.C.; Elleder, M.; Karetova, D.; Pavlikova, M.; Hrebicek, M. Relationship between X-inactivation and clinical involvement in Fabry heterozygotes. Eleven novel mutations in the α-galactosidase a gene in the Czech and Slovak population. J. Mol. Med. 2005, 83, 647–654. [Google Scholar] [CrossRef]
- Beck, M.; Cox, T.M. Comment: Why are females with Fabry disease affected? Mol. Genet. Metab. Rep. 2019, 21, 100529. [Google Scholar] [CrossRef]
- Wilcox, W.R.; Oliveira, J.P.; Hopkin, R.J.; Ortiz, A.; Banikazemi, M.; Feldt-Rasmussen, U.; Sims, K.; Waldek, S.; Pastores, G.M.; Lee, P.; et al. Females with Fabry disease frequently have major organ involvement: Lessons from the Fabry Registry. Mol. Genet. Metab. 2008, 93, 112–128. [Google Scholar] [CrossRef] [PubMed]
- Gal, A.; Hughes, D.A.; Winchester, B. Toward a consensus in the laboratory diagnostics of Fabry disease - recommendations of a European expert group. J. Inherit. Metab. Dis. 2011, 34, 509–514. [Google Scholar] [CrossRef] [Green Version]
- Massaccesi, L.; Burlina, A.; Baquero, C.J.; Goi, G.; Burlina, A.P.; Tettamanti, G. Whole-blood α-D-galactosidase A activity for the identification of Fabry’s patients. Clin. Biochem. 2011, 44, 916–921. [Google Scholar] [CrossRef]
- Hölzl, M.A.; Gärtner, M.; Kovarik, J.J.; Hofer, J.; Bernheimer, H.; Sunder-Plassmann, G.; Zlabinger, G.J. Quantification of α-galactosidase activity in intact leukocytes. Clin. Chim. Acta 2010, 411, 1666–1670. [Google Scholar] [CrossRef]
- Linthorst, G.E.; Vedder, A.C.; Aerts, J.M.F.G.; Hollak, C.E.M. Screening for Fabry disease using whole blood spots fails to identify one-third of female carriers. Clin. Chim. Acta 2005, 353, 201–203. [Google Scholar] [CrossRef]
- Trimarchi, H.; Canzonieri, R.; Schiel, A.; Costales-Collaguazo, C.; Politei, J.; Stern, A.; Paulero, M.; Rengel, T.; Andrews, J.; Forrester, M.; et al. Increased urinary CD80 excretion and podocyturia in Fabry disease. J. Transl. Med. 2016, 14, 289. [Google Scholar] [CrossRef] [Green Version]
- Vedder, A.C.; Linthorst, G.E.; van Breemen, M.J.; Groener, J.E.M.; Bemelman, F.J.; Strijland, A.; Mannens, M.M.A.M.; Aerts, J.M.F.G.; Hollak, C.E.M. The Dutch Fabry cohort: Diversity of clinical manifestations and Gb3 levels. J. Inherit. Metab. Dis. 2007, 30, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Bekri, S.; Lidove, O.; Jaussaud, R.; Knebelmann, B.; Barbey, F. The role of ceramide trihexoside (globotriaosylceramide) in the diagnosis and follow-up of the efficacy of treatment of Fabry Disease: A review of the literature. Cardiovasc. Hematol. Agents Med. Chem. 2008, 4, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Nowak, A.; Mechtler, T.P.; Desnick, R.J.; Kasper, D.C. Plasma LysoGb3: A useful biomarker for the diagnosis and treatment of Fabry disease heterozygotes. Mol. Genet. Metab. 2017, 120, 57–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Togawa, T.; Kodama, T.; Suzuki, T.; Sugawara, K.; Tsukimura, T.; Ohashi, T.; Ishige, N.; Suzuki, K.; Kitagawa, T.; Sakuraba, H. Plasma globotriaosylsphingosine as a biomarker of Fabry disease. Mol. Genet. Metab. 2010, 100, 257–261. [Google Scholar] [CrossRef]
- Sakuraba, H.; Togawa, T.; Tsukimura, T.; Kato, H. Plasma lyso-Gb3: A biomarker for monitoring fabry patients during enzyme replacement therapy. Clin. Exp. Nephrol. 2018, 22, 843–849. [Google Scholar] [CrossRef] [Green Version]
- Arends, M.; Biegstraaten, M.; Wanner, C.; Sirrs, S.; Mehta, A.; Elliott, P.M.; Oder, D.; Watkinson, O.T.; Bichet, D.G.; Khan, A.; et al. Agalsidase alfa versus agalsidase β for the treatment of Fabry disease: An international cohort study. J. Med. Genet. 2018, 55, 351–358. [Google Scholar] [CrossRef] [Green Version]
- Ortiz, A.; Germain, D.P.; Desnick, R.J.; Politei, J.; Mauer, M.; Burlina, A.; Eng, C.; Hopkin, R.J.; Laney, D.; Linhart, A.; et al. Fabry disease revisited: Management and treatment recommendations for adult patients. Mol. Genet. Metab. 2018, 123, 416–427. [Google Scholar] [CrossRef]
- Fan, J.Q.; Ishii, S. Active-site-specific chaperone therapy for Fabry disease. Yin and Yang of enzyme inhibitors. FEBS J. 2007, 274, 4962–4971. [Google Scholar] [CrossRef]
- Ortiz, A.; Oliveira, J.P.; Waldek, S.; Warnock, D.G.; Cianciaruso, B.; Wanner, C. Nephropathy in males and females with Fabry disease: Cross-sectional description of patients before treatment with enzyme replacement therapy. Nephrol. Dial. Transplant. 2008, 23, 1600–1607. [Google Scholar] [CrossRef] [Green Version]
- Pisani, A.; Visciano, B.; Imbriaco, M.; Di Nuzzi, A.; Mancini, A.; Marchetiello, C.; Riccio, E. The kidney in Fabry’s disease. Clin. Genet. 2014, 86, 301–309. [Google Scholar] [CrossRef] [Green Version]
- Ortiz, A.; Oliveira, J.P.; Wanner, C.; Brenner, B.M.; Waldek, S.; Warnock, D.G. Recommendations and guidelines for the diagnosis and treatment of Fabry nephropathy in adults. Nat. Clin. Pract. Nephrol. 2008, 4, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Alroy, J.; Sabnis, S.; Kopp, J.B. Renal pathology in Fabry disease. J. Am. Soc. Nephrol. 2002, 13, S134–S138. [Google Scholar] [CrossRef]
- Vujkovac, B. Fabry disease: Diagnostic methods in nephrology practice. Clin. Nephrol. 2017, 88, 44–47. [Google Scholar] [CrossRef]
- Tøndel, C.; Bostad, L.; Hirth, A.; Svarstad, E. Renal biopsy findings in children and adolescents with Fabry disease and minimal albuminuria. Am. J. Kidney Dis. 2008, 51, 767–776. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.M.; Nikolic-Paterson, D.J.; Lan, H.Y. Inflammatory processes in renal fibrosis. Nat. Rev. Nephrol. 2014, 10, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Liu, Y. Renal fibrosis in 2015: Understanding the mechanisms of kidney fibrosis. Nat. Rev. Nephrol. 2016, 12, 68–70. [Google Scholar] [CrossRef]
- Levey, A.S.; Stevens, L.A.; Schmid, C.H.; Zhang, Y.; Castro, A.F.; Feldman, H.I.; Kusek, J.W.; Eggers, P.; Van Lente, F.; Greene, T.; et al. A new equation to estimate glomerular filtration rate. Ann. Intern. Med. 2009, 150, 604–612. [Google Scholar] [CrossRef]
- Schwartz, G.J.; Muñoz, A.; Schneider, M.F.; Mak, R.H.; Kaskel, F.; Warady, B.A.; Furth, S.L. New equations to estimate GFR in children with CKD. J. Am. Soc. Nephrol. 2009, 20, 629–637. [Google Scholar] [CrossRef] [Green Version]
- Riccio, E.; Sabbatini, M.; Bruzzese, D.; Annicchiarico Petruzzelli, L.; Pellegrino, A.; Spinelli, L.; Esposito, R.; Imbriaco, M.; Feriozzi, S.; Pisani, A. Glomerular hyperfiltration: An early marker of nephropathy in Fabry disease. Nephron 2019, 141, 10–17. [Google Scholar] [CrossRef]
- Ramaswami, U.; Najafian, B.; Schieppati, A.; Mauer, M.; Bichet, D.G. Assessment of renal pathology and dysfunction in pediatric patients with Fabry disease. Clin. J. Am. Soc. Nephrol. 2010, 5, 365–370. [Google Scholar] [CrossRef] [Green Version]
- Schiffmann, R.; Warnock, D.G.; Banikazemi, M.; Bultas, J.; Linthorst, G.E.; Packman, S.; Sorensen, S.A.; Wilcox, W.R.; Desnick, R.J. Fabry disease: Progression of nephropathy, and prevalence of cardiac and cerebrovascular events before enzyme replacement therapy. Nephrol. Dial. Transplant. 2009, 24, 2102–2111. [Google Scholar] [CrossRef] [PubMed]
- Torralba-Cabeza, M.Á.; Olivera, S.; Hughes, D.A.; Pastores, G.M.; Mateo, R.N.; Pérez-Calvo, J.I. Cystatin C and NT-proBNP as prognostic biomarkers in Fabry disease. Mol. Genet. Metab. 2011, 104, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Gupta, P.; Pyeritz, R.E.; Kwiterovich, P.O.J. Immunohistochemical localization of glycosphingolipid in urinary renal tubular cells in Fabry’s disease. Am. J. Clin. Pathol 1984, 82, 24–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagao, S.; Satoh, N.; Inaba, S.; Iijima, S. Concentric lamellar spheres in urine from a female carrier of and patients with Fabry’s disease—With special reference to polarization and electron microscopic comparison with nephrotic syndrome. J. Dermatol. 1985, 12, 70–78. [Google Scholar] [CrossRef]
- Desnick, R.J.; Dawson, G.; Desnick, S.J.; Sweeley, C.C.; Krivit, W. Diagnosis of glycosphingolipidoses by urinary-sediment analysis. N. Engl. J. Med. 1971, 284, 739–744. [Google Scholar] [CrossRef]
- Selvarajah, M.; Nicholls, K.; Hewitson, T.D.; Becker, G.J. Targeted urine microscopy in Anderson-Fabry disease: A cheap, sensitive and specific diagnostic technique. Nephrol. Dial. Transplant. 2011, 26, 3195–3202. [Google Scholar] [CrossRef]
- van der Tol, L.; Svarstad, E.; Ortiz, A.; Tøndel, C.; Oliveira, J.P.; Vogt, L.; Waldek, S.; Hughes, D.A.; Lachmann, R.H.; Terryn, W.; et al. Chronic kidney disease and an uncertain diagnosis of Fabry disease: Approach to a correct diagnosis. Mol. Genet. Metab. 2015, 114, 242–247. [Google Scholar] [CrossRef]
- Skrunes, R.; Tøndel, C.; Leh, S.; Larsen, K.K.; Houge, G.; Davidsen, E.S.; Hollak, C.; Van Kuilenburg, A.B.P.; Vaz, F.M.; Svarstad, E. Long-term dose-dependent agalsidase effects on kidney histology in Fabry disease. Clin. J. Am. Soc. Nephrol. 2017, 12, 1470–1479. [Google Scholar] [CrossRef] [Green Version]
- Fogo, A.B.; Bostad, L.; Svarstad, E.; Cook, W.J.; Moll, S.; Barbey, F.; Geldenhuys, L.; West, M.; Ferluga, D.; Vujkovac, B.; et al. Scoring system for renal pathology in Fabry disease: Report of the International Study Group of Fabry Nephropathy (ISGFN). Nephrol. Dial. Transplant. 2010, 25, 2168–2177. [Google Scholar] [CrossRef]
- Ramaswami, U.; Bichet, D.G.; Clarke, L.A.; Dostalova, G.; Fainboim, A.; Fellgiebel, A.; Forcelini, C.M.; An Haack, K.; Hopkin, R.J.; Mauer, M.; et al. Low-dose agalsidase β treatment in male pediatric patients with Fabry disease: A 5-year randomized controlled trial. Mol. Genet. Metab. 2019, 127, 86–94. [Google Scholar] [CrossRef]
- Najafian, B.; Svarstad, E.; Bostad, L.; Gubler, M.C.; Tøndel, C.; Whitley, C.; Mauer, M. Progressive podocyte injury and globotriaosylceramide (GL-3) accumulation in young patients with Fabry disease. Kidney Int. 2011, 79, 663–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valbuena, C.; Carvalho, E.; Bustorff, M.; Ganhão, M.; Relvas, S.; Nogueira, R.; Carneiro, F.; Oliveira, J.P. Kidney biopsy findings in heterozygous Fabry disease females with early nephropathy. Virchows Arch. 2008, 453, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Fischer, E.G.; Moore, M.J.; Lager, D.J. Fabry disease: A morphologic study of 11 cases. Mod. Pathol. 2006, 19, 1295–1301. [Google Scholar] [CrossRef] [PubMed]
- Svarstad, E.; Leh, S.; Skrunes, R.; Kampevold Larsen, K.; Eikrem, Ø.; Tøndel, C. Bedside stereomicroscopy of Fabry kidney biopsies: An easily available method for diagnosis and assessment of sphingolipid deposits. Nephron 2018, 138, 13–21. [Google Scholar] [CrossRef]
- Thurberg, B.L.; Rennke, H.; Colvin, R.B.; Dikman, S.; Gordon, R.E.; Collins, A.B.; Desnick, R.J.; O’Callaghan, M. Globotriaosylceramide accumulation in the Fabry kidney is cleared from multiple cell types after enzyme replacement therapy. Kidney Int. 2002, 62, 1933–1946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Germain, D.P.; Waldek, S.; Banikazemi, M.; Bushinsky, D.A.; Charrow, J.; Desnick, R.J.; Lee, P.; Loew, T.; Vedder, A.C.; Abichandani, R.; et al. Sustained, long-term renal stabilization after 54 months of agalsidase β therapy in patients with Fabry disease. J. Am. Soc. Nephrol. 2007, 18, 1547–1557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramaswami, U.; Beck, M.; Hughes, D.; Kampmann, C.; Botha, J.; Pintos-Morell, G.; West, M.L.; Niu, D.M.; Nicholls, K.; Giugliani, R. Cardio-renal outcomes with long-term agalsidase alfa enzyme replacement therapy: A 10-year Fabry Outcome Survey (FOS) analysis. Drug Des. Devel. Ther. 2019, 13, 3705–3715. [Google Scholar] [CrossRef] [Green Version]
- Wanner, C.; Feldt-Rasmussen, U.; Jovanovic, A.; Linhart, A.; Yang, M.; Ponce, E.; Brand, E.; Germain, D.P.; Hughes, D.A.; Jefferies, J.L.; et al. Cardiomyopathy and kidney function in agalsidase β-treated female Fabry patients: A pre-treatment vs. post-treatment analysis. ESC Hear. Fail. 2020, 7, 825–834. [Google Scholar] [CrossRef]
- Schiffmann, R.; Askari, H.; Timmons, M.; Robinson, C.; Benko, W.; Brady, R.O.; Ries, M. Weekly enzyme replacement therapy may slow decline of renal function in patients with Fabry disease who are on long-term biweekly dosing. J. Am. Soc. Nephrol 2007, 18, 1576–1583. [Google Scholar] [CrossRef]
- Wanner, C.; Oliveira, J.P.; Ortiz, A.; Mauer, M.; Germain, D.P.; Linthorst, G.E.; Serra, A.L.; Maródi, L.; Mignani, R.; Cianciaruso, B.; et al. Prognostic indicators of renal disease progression in adults with fabry disease: Natural history data from the Fabry Registry. Clin. J. Am. Soc. Nephrol. 2010, 5, 2220–2228. [Google Scholar] [CrossRef]
- Taal, M.W.; Brenner, B.M. Predicting initiation and progression of chronic kidney disease: Developing renal risk scores. Kidney Int. 2006, 70, 1694–1705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verovnik, F.; Benko, D.; Vujkovac, B.; Linthorst, G.E. Remarkable variability in renal disease in a large Slovenian family with Fabry disease. Eur. J. Hum. Genet. 2004, 12, 678–681. [Google Scholar] [CrossRef] [PubMed]
- Lepedda, A.J.; Fancellu, L.; Zinellu, E.; De Muro, P.; Nieddu, G.; Deiana, G.A.; Canu, P.; Concolino, D.; Sestito, S.; Formato, M.; et al. Urine bikunin as a marker of renal impairment in Fabry’s disease. Biomed. Res. Int. 2013, 2013, 205948. [Google Scholar] [CrossRef] [PubMed]
- Aguiar, P.; Azevedo, O.; Pinto, R.; Marino, J.; Baker, R.; Cardoso, C.; Ducla Soares, J.L.; Hughes, D. New biomarkers defining a novel early stage of Fabry nephropathy: A diagnostic test study. Mol. Genet. Metab. 2017, 121, 162–169. [Google Scholar] [CrossRef]
- Schiffmann, R.; Waldek, S.; Benigni, A.; Auray-Blais, C. Biomarkers of Fabry disease nephropathy. Clin. J. Am. Soc. Nephrol. 2010, 5, 360–364. [Google Scholar] [CrossRef] [PubMed]
- Prabakaran, T.; Birn, H.; Bibby, B.M.; Regeniter, A.; Sørensen, S.S.; Feldt-Rasmussen, U.; Nielsen, R.; Christensen, E.I. Long-term enzyme replacement therapy is associated with reduced proteinuria and preserved proximal tubular function in women with Fabry disease. Nephrol. Dial. Transplant. 2014, 29, 619–625. [Google Scholar] [CrossRef]
- Vylet’al, P.; Hålková, H.; Živná, M.; Berná, L.; Novák, P.; Elleder, M.; Kmoch, S. Abnormal expression and processing of uromodulin in Fabry disease reflects tubular cell storage alteration and is reversible by enzyme replacement therapy. J. Inherit. Metab. Dis. 2008, 31, 508–517. [Google Scholar] [CrossRef] [Green Version]
- Tøndel, C.; Kanai, T.; Larsen, K.K.; Ito, S.; Politei, J.M.; Warnock, D.G.; Svarstad, E. Foot process effacement is an early marker of nephropathy in young classic Fabry patients without albuminuria. Nephron 2015, 129, 16–21. [Google Scholar] [CrossRef]
- Fall, B.; Scott, C.R.; Mauer, M.; Shankland, S.; Pippin, J.; Jefferson, J.A.; Wallace, E.; Warnock, D.; Najafian, B. Urinary podocyte loss is increased in patients with Fabry disease and correlates with clinical severity of Fabry nephropathy. PLoS ONE 2016, 11, e0168346. [Google Scholar] [CrossRef] [Green Version]
- Najafian, B.; Tøndel, C.; Svarstad, E.; Gubler, M.C.; Oliveira, J.P.; Mauer, M. Accumulation of globotriaosylceramide in podocytes in Fabry nephropathy is associated with progressive podocyte loss. J. Am. Soc. Nephrol. 2020, 31, 865–875. [Google Scholar] [CrossRef]
- Trimarchi, H.; Canzonieri, R.; Costales-Collaguazo, C.; Politei, J.; Stern, A.; Paulero, M.; González-Hoyos, I.; Schiel, A.; Rengel, T.; Forrester, M.; et al. Early decrease in the podocalyxin to synaptopodin ratio in urinary Fabry podocytes. Clin. Kidney J. 2019, 12, 53–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- May, C.J.; Saleem, M.; Welsh, G.I. Podocyte dedifferentiation: A specialized process for a specialized cell. Front. Endocrinol. 2014, 5, 148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weidemann, F.; Sanchez-Niño, M.D.; Politei, J.; Oliveira, J.P.; Wanner, C.; Warnock, D.G.; Ortiz, A. Fibrosis: A key feature of Fabry disease with potential therapeutic implications. Orphanet J. Rare Dis. 2013, 8, 116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, E.M.; da Silva, A.S.; Labilloy, A.; do Monte Neto, J.T.; do Monte, S.J.H. Podocyturia in Fabry disease. J. Bras. Nefrol. 2016, 38, 49–53. [Google Scholar] [CrossRef]
- Altarescu, G.; Chicco, G.; Whybra, C.; Delgado-Sanchez, S.; Sharon, N.; Beck, M.; Elstein, D. Correlation between interleukin-6 promoter and C-reactive protein (CRP) polymorphisms and CRP levels with the Mainz Severity Score Index for Fabry disease. J. Inherit. Metab. Dis. 2008, 31, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Altarescu, G.; Moore, D.F.; Schiffmann, R. Effect of genetic modifiers on cerebral lesions in Fabry disease. Neurology 2005, 64, 2148–2150. [Google Scholar] [CrossRef]
- Heltianu, C.; Costache, G.; Azibi, K.; Poenaru, L.; Simionescu, M. Endothelial nitric oxide synthase gene polymorphisms in Fabry’s disease. Clin. Genet. 2002, 61, 423–429. [Google Scholar] [CrossRef]
- Scionti, F.; Di Martino, M.T.; Sestito, S.; Nicoletti, A.; Falvo, F.; Roppa, K.; Arbitrio, M.; Guzzi, P.H.; Agapito, G.; Pisani, A.; et al. Genetic variants associated with Fabry disease progression despite enzyme replacement therapy. Oncotarget 2017, 8, 107558–107564. [Google Scholar] [CrossRef] [Green Version]
- Jaurretche, S.; Perez, G.R.; Venera, G. High Lyso-Gb3 plasma levels associated with decreased miR-29 and miR-200 urinary excretion in young non-albuminuric male patient with classic Fabry disease. Case Rep. Nephrol. 2019, 2019, 4980942. [Google Scholar] [CrossRef] [Green Version]
- Jaurretche, S.; Venera, G.; Antongiovanni, N.; Perretta, F.; Perez, G.R. Urinary excretion of microRNAs in young Fabry disease patients with mild or absent nephropathy. Open J. Nephrol. 2018, 8, 71–83. [Google Scholar] [CrossRef] [Green Version]
- Xiao, K.; Lu, D.; Hoepfner, J.; Santer, L.; Gupta, S.; Pfanne, A.; Thum, S.; Lenders, M.; Brand, E.; Nordbeck, P.; et al. Circulating microRNAs in Fabry disease. Sci. Rep. 2019, 9, 15277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Condrat, C.E.; Thompson, D.C.; Barbu, M.G.; Bugnar, O.L.; Boboc, A.; Cretoiu, D.; Suciu, N.; Cretoiu, S.M.; Voinea, S.C. miRNAs as Biomarkers in Disease: Latest Findings Regarding Their Role in Diagnosis and Prognosis. Cells 2020, 9, 276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hübner, A.; Metz, T.; Schanzer, A.; Greber-Platzer, S.; Item, C.B. Aberrant DNA methylation of calcitonin receptor in Fabry patients treated with enzyme replacement therapy. Mol. Genet. Metab. Rep. 2015, 5, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.A.; Wu, C.; Yanagisawa, H.; Miyajima, T.; Akiyama, K.; Eto, Y. Future clinical and biochemical predictions of Fabry disease in females by methylation studies of the GLA gene. Mol. Genet. Metab. Rep. 2019, 20, 100497. [Google Scholar] [CrossRef] [PubMed]
- Abaoui, M.; Boutin, M.; Lavoie, P.; Auray-Blais, C. Tandem mass spectrometry multiplex analysis of methylated and non-methylated urinary Gb3 isoforms in Fabry disease patients. Clin. Chim. Acta 2016, 452, 191–198. [Google Scholar] [CrossRef]
- Chandramouli, K.; Qian, P.-Y. Proteomics: Challenges, Techniques and Possibilities to Overcome Biological Sample Complexity. Hum. Genom. Proteom. 2009, 1, 239204. [Google Scholar] [CrossRef] [Green Version]
- Johnson, C.H.; Gonzalez, F.J. Challenges and Opportunities of Metabolomics. J. Cell Physiol. 2012, 227, 2975–2981. [Google Scholar] [CrossRef]
- Auray-Blais, C.; Boutin, M.; Gagnon, R.; Dupont, F.O.; Lavoie, P.; Clarke, J.T.R. Urinary globotriaosylsphingosine-related biomarkers for Fabry disease targeted by metabolomics. Anal. Chem. 2012, 84, 2745–2753. [Google Scholar] [CrossRef]
- Manwaring, V.; Boutin, M.; Auray-Blais, C. A metabolomic study to identify new globotriaosylceramide-related biomarkers in the plasma of Fabry disease patients. Anal. Chem. 2013, 85, 9039–9048. [Google Scholar] [CrossRef] [PubMed]
- Dupont, F.O.; Gagnon, R.; Boutin, M.; Auray-Blais, C. A metabolomic study reveals novel plasma lyso-Gb3 analogs as Fabry disease biomarkers. Curr. Med. Chem. 2013, 20, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Niño, M.D.; Sanz, A.B.; Carrasco, S.; Saleem, M.A.; Mathieson, P.W.; Valdivielso, J.M.; Ruiz-Ortega, M.; Egido, J.; Ortiz, A. Globotriaosylsphingosine actions on human glomerular podocytes: Implications for Fabry nephropathy. Nephrol. Dial. Transplant. 2011, 26, 1797–1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rozenfeld, P.; Feriozzi, S. Contribution of inflammatory pathways to Fabry disease pathogenesis. Mol. Genet. Metab. 2017, 122, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Trimarchi, H. Abatacept and glomerular diseases: The open road for the second signal as a new target is settled down. Recent Pat. Endocr. Metab. Immune Drug Discov. 2015, 9, 2–14. [Google Scholar] [CrossRef] [PubMed]
- Sutariya, B.; Jhonsa, D.; Saraf, M.N. TGF-β: The connecting link between nephropathy and fibrosis. Immunopharmacol. Immunotoxicol. 2016, 38, 39–49. [Google Scholar] [CrossRef]
- Sanchez-Niño, M.D.; Sanz, A.B.; Ruiz-Andres, O.; Poveda, J.; Izquierdo, M.C.; Selgas, R.; Egido, J.; Ortiz, A. MIF, CD74 and other partners in kidney disease: Tales of a promiscuous couple. Cytokine Growth Factor Rev. 2013, 24, 23–40. [Google Scholar] [CrossRef] [PubMed]
- Auray-Blais, C.; Ntwari, A.; Clarke, J.T.R.; Warnock, D.G.; Oliveira, J.P.; Young, S.P.; Millington, D.S.; Bichet, D.G.; Sirrs, S.; West, M.L.; et al. How well does urinary lyso-Gb3 function as a biomarker in Fabry disease? Clin. Chim. Acta 2010, 411, 1906–1914. [Google Scholar] [CrossRef] [PubMed]
- Boutin, M.; Auray-Blais, C. Multiplex tandem mass spectrometry analysis of novel plasma lyso-Gb3-related analogues in Fabry disease. Anal. Chem. 2014, 86, 3476–3483. [Google Scholar] [CrossRef]
- Alharbi, F.J.; Baig, S.; Rambhatla, S.B.; Vijapurapu, R.; Auray-Blais, C.; Boutin, M.; Steeds, R.; Wheeldon, N.; Dawson, C.; Geberhiwot, T. The clinical utility of total concentration of urinary globotriaosylsphingosine plus its analogues in the diagnosis of Fabry disease. Clin. Chim. Acta 2020, 500, 120–127. [Google Scholar] [CrossRef]
- Boutin, M.; Auray-Blais, C. Metabolomic discovery of novel urinary galabiosylceramide analogs as Fabry disease biomarkers. J. Am. Soc. Mass Spectrom. 2015, 26, 499–510. [Google Scholar] [CrossRef] [Green Version]
- Auray-Blais, C.; Blais, C.M.; Ramaswami, U.; Boutin, M.; Germain, D.P.; Dyack, S.; Bodamer, O.; Pintos-Morell, G.; Clarke, J.T.R.; Bichet, D.G.; et al. Urinary biomarker investigation in children with Fabry disease using tandem mass spectrometry. Clin. Chim. Acta 2014, 438, 195–204. [Google Scholar] [CrossRef]
- Tebani, A.; Mauhin, W.; Abily-Donval, L.; Lesueur, C.; Berger, M.G.; Nadjar, Y.; Berger, J.; Benveniste, O.; Lamari, F.; Laforêt, P.; et al. A proteomics-based analysis reveals predictive biological patterns in Fabry disease. J. Clin. Med. 2020, 9, 1325. [Google Scholar] [CrossRef] [PubMed]
- Doykov, I.D.; Heywood, W.E.; Nikolaenko, V.; Å Piewak, J.; Hällqvist, J.; Clayton, P.T.; Mills, P.; Warnock, D.G.; Nowak, A.; Mills, K. Rapid, proteomic urine assay for monitoring progressive organ disease in Fabry disease. J. Med. Genet. 2020, 57, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Slaats, G.G.; Braun, F.; Hoehne, M.; Frech, L.E.; Blomberg, L.; Benzing, T.; Schermer, B.; Rinschen, M.M.; Kurschat, C.E. Urine-derived cells: A promising diagnostic tool in Fabry disease patients. Sci. Rep. 2018, 8, 11042. [Google Scholar] [CrossRef] [PubMed]
- Heo, S.H.; Kang, E.; Kim, Y.M.; Go, H.; Kim, K.Y.; Jung, J.Y.; Kang, M.; Kim, G.H.; Kim, J.M.; Choi, I.H.; et al. Fabry disease: Characterisation of the plasma proteome pre- and post-enzyme replacement therapy. J. Med. Genet. 2017, 54, 771–780. [Google Scholar] [CrossRef]
- Matafora, V.; Cuccurullo, M.; Beneduci, A.; Petrazzuolo, O.; Simeone, A.; Anastasio, P.; Mignani, R.; Feriozzi, S.; Pisani, A.; Comotti, C.; et al. Early markers of Fabry disease revealed by proteomics. Mol. Biosyst. 2015, 11, 1543–1551. [Google Scholar] [CrossRef]
- Hollander, Z.; Dai, D.L.Y.; Putko, B.N.; Yogasundaram, H.; Wilson-Mcmanus, J.E.; Thompson, R.B.; Khan, A.; West, M.L.; McManus, B.M.; Oudit, G.Y. Gender-specific plasma proteomic biomarkers in patients with Anderson-Fabry disease. Eur. J. Heart Fail. 2015, 17, 291–300. [Google Scholar] [CrossRef] [Green Version]
- Cigna, D.; D’Anna, C.; Zizzo, C.; Francofonte, D.; Sorrentino, I.; Colomba, P.; Albeggiani, G.; Armini, A.; Bianchi, L.; Bini, L.; et al. Alteration of proteomic profiles in PBMC isolated from patients with Fabry disease: Preliminary findings. Mol. Biosyst. 2013, 9, 1162–1168. [Google Scholar] [CrossRef]
- Manwaring, V.; Heywood, W.E.; Clayton, R.; Lachmann, R.H.; Keutzer, J.; Hindmarsh, P.; Winchester, B.; Heales, S.; Mills, K. The identification of new biomarkers for identifying and monitoring kidney disease and their translation into a rapid mass spectrometry-based test: Evidence of presymptomatic kidney disease in pediatric Fabry and type-I diabetic patients. J. Proteome Res. 2013, 12, 2013–2021. [Google Scholar] [CrossRef]
- Kistler, A.D.; Siwy, J.; Breunig, F.; Jeevaratnam, P.; Scherl, A.; Mullen, W.; Warnock, D.G.; Wanner, C.; Hughes, D.A.; Mischak, H.; et al. A distinct urinary biomarker pattern characteristic of female Fabry patients that mirrors response to enzyme replacement therapy. PLoS ONE 2011, 6, e20534. [Google Scholar] [CrossRef]
- Vojtová, L.; Zima, T.; Tesař, V.; Michalová, J.; Přikryl, P.; Dostálová, G.; Linhart, A. Study of urinary proteomes in Anderson-Fabry disease. Ren. Fail. 2010, 32, 1202–1209. [Google Scholar] [CrossRef]
- Moore, D.F.; Krokhin, O.V.; Beavis, R.C.; Ries, M.; Robinson, C.; Goldin, E.; Brady, R.O.; Wilkins, J.A.; Schiffmann, R. Proteomics of specific treatment-related alterations in Fabry disease: A strategy to identify biological abnormalities. Proc. Natl. Acad. Sci. USA 2007, 104, 2873–2878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heywood, W.E.; Doykov, I.; Spiewak, J.; Hallqvist, J.; Mills, K.; Nowak, A. Global glycosphingolipid analysis in urine and plasma of female Fabry disease patients. Biochim. Biophys. Acta—Mol. Basis Dis. 2019, 1865, 2726–2735. [Google Scholar] [CrossRef] [PubMed]
- Boutin, M.; Menkovic, I.; Martineau, T.; Vaillancourt-Lavigueur, V.; Toupin, A.; Auray-Blais, C. Separation and analysis of lactosylceramide, G galabiosylceramide, and globotriaosylceramide by LC-MS/MS in urine of Fabry disease patients. Anal. Chem. 2017, 89, 13382–13390. [Google Scholar] [CrossRef] [PubMed]
- Boutin, M.; Lavoie, P.; Abaoui, M.; Auray-Blais, C. Tandem mass spectrometry quantitation of lyso-Gb3 and six related analogs in plasma for Fabry disease patients. Curr. Protoc. Hum. Genet. 2016, 90, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Sueoka, H.; Ichihara, J.; Tsukimura, T.; Togawa, T.; Sakuraba, H. Nano-LC-MS/MS for quantification of lyso-Gb3 and its analogues reveals a useful biomarker for Fabry disease. PLoS ONE 2015, 10, e0127048. [Google Scholar] [CrossRef] [PubMed]
- Lavoie, P.; Boutin, M.; Auray-Blais, C. Multiplex analysis of novel urinary lyso-Gb3-related biomarkers for Fabry disease by tandem mass spectrometry. Anal. Chem. 2013, 85, 1743–1752. [Google Scholar] [CrossRef]
- Auray-Blais, C.; Boutin, M. Novel Gb3 isoforms detected in urine of Fabry disease patients: A metabolomic study. Curr. Med. Chem. 2012, 19, 3241–3252. [Google Scholar] [CrossRef]
- Colhoun, H.M.; Marcovecchio, M.L. Biomarkers of diabetic kidney disease. Diabetologia 2018, 61, 996–1011. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.H.; Bang, H.; Kim, D.J. How to establish clinical prediction models. Endocrinol. Metab. 2016, 31, 38–44. [Google Scholar] [CrossRef]
- Tangri, N.; Stevens, L.A.; Griffith, J.; Tighiouart, H.; Djurdjev, O.; Naimark, D.; Levin, A.; Levey, A.S. A predictive model for progression of chronic kidney disease to kidney failure. JAMA—J. Am. Med. Assoc. 2011, 305, 1553–1559. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Subjects | Sample Type | Methods Used | Outcomes in FD | Reference |
|---|---|---|---|---|
| 69 FD patients and 83 healthy subjects | plasma | electrochemi-luminescence assays | FD patients: elevation of FGF2, VEGFA, VEGFC and IL-7. Non-classical FD patients: significant correlation between IL-7 and residual enzyme activity. | [88] |
| 66 FD patients and 10 healthy subjects | urine | LC-MS/MS | Early stage and asymptomatic FD patients: elevation of albumin, uromodulin, α1-antitrypsin, glycogen phosphorylase brain form, endothelial protein receptor 1 and intracellular adhesion molecule 1. | [89] |
| 7 FD patients and 7 healthy controls | urine-derived cells | Label-free quantitative nLC-MS/MS | 46 candidate proteins with altered expression indicating an involvement of the Coordinated Lysosomal Expression and Regulation (CLEAR). | [90] |
| 8 classic FD patients | plasma | 2D electro-phoresis and MALDI-TOF-MS and MS/MS | Short-term ERT: reduction of 15 plasma proteins involved in inflammation, oxidative and ischemic injury, or complement activation. Pre-ERT: significant elevation of plasma β-actin, inactivated complement C3b (iC3b), and C4B. Longer-term ERT: gradually decreased iC3b. | [91] |
| 11 naïve Fabry patients, 12 permanently ERT-treated patients and 12 healthy subjects | second morning urine | free approach coupled with LC-MS/MS analysis | Up-regulation: uromodulin, prostaglandin H2 D-isomerase and prosaposin. | [92] |
| 32 FD patients and 14 healthy subjects | plasma | MS iTRAQ followed by MRM approach | Male FD patients: specific and sensitive panel of eight proteins (22 kDa protein, afamin, 𝛼1 antichymotrypsin, apolipoprotein E, 𝛽-Ala His dipeptidase, hemoglobin 𝛼-2, isoform 1 of sex hormone-binding globulin, and peroxiredoxin 2). Female FD patients: a nine-protein marker panel; where three proteins (apolipoprotein E, hemoglobin 𝛼-2, and peroxiredoxin 2) were common to both genders. | [93] |
| 8 FD and 6 healthy subjects | PBMC | 2D electro-phoresis and MALDI-TOF-MS | Downregulated: calnexin, rho GDP dissociation inhibitor 2, rho GDP-dissociation inhibitor 1, chloride intracellular channel protein 1. Up-regulated: g-enolase, 14-3-3 protein theta, 14-3-3 protein zeta/delta, and galectin-1. | [94] |
| 10 male pediatric FD patients | urine | QTOF-MS | Prosaposin: significantly elevated in FD in and reduced after 12 months of ERT. GM2AP: elevated in the pretreatment Fabry patients and reduced after 12 months of ERT. | [95] |
| 35 treatment-naïve female FD patients, 89 healthy subjects | urine | CE-MS | Urinary biomarker profile for female patients: diagnostics of the FD, monitoring of ERT. | [96] |
| 20 FD patients, 10 healthy controls | urine | SDS-PAGE and MALDI-TOF MS | Differentially expressed proteins: α-1-antitrypsin, α-1-microglobulin, prostaglandin H2 D-isomerase, complement-c1q tumor necrosis factor-related protein, and Ig kappa chain V–III. | [97] |
| 13 pediatric FD patients | serum | LC-MS/MS | Reduced after 6 months of ERT: α2-HS glycoprotein, vitamin D-binding protein, transferrin, Ig- α-2 C chain, and α-2-antiplasmin. | [98] |
| Subjects | Sample Type | Methods Used | Outcomes | Reference |
|---|---|---|---|---|
| 42 FD patients, 48 healthy controls | random urine | LC-MS/MS | The total urinary concentration of lyso-Gb3 and its analogues: 100% specific for classical and non-classical FD patients. | [110] |
| 18 asymptomatic females, 18 symptomatic females, 27 males, 16 control urines, 58 control plasmas | plasma and urine | UPLC-MS/MS | Urinary ceramide dihexoside (CDH):
| [113] |
| 34 untreated and 33 treated Fabry males, 54 untreated and 19 treated females, 34 males and 25 female healthy control | random urine | UPLC-MS/MS | Validated method for separation of 12 most abundant Ga2 isoforms/analogs from their lactosylceramide (LacCer) counterparts:
| [114] |
| 15 untreated and 28 treated Fabry males, 21 untreated and 10 treated Fabry females, 15 males and 26 female healthy control | plasma | UPLC-MS/MS | Validated UPLC-MS/MS method for the multiplex analysis of lyso-Gb3 and its 6 analogs. | [115] |
| 16 untreated Fabry males, 16 healthy Fabry males | random urine | TOF MS | Untreated FD patients: 22 urinary Ga2 isoforms/analogs; quantification of Ga2 and Gb3 urinary isoforms/analogs that were elevated in untreated Fabry males. | [111] |
| 9 males with classic FD, 7 males with later-onset FD, 10 females, 5 males with functional variants, 40 healthy controls | plasma | nano-LC-MS/MS | Plasma lyso-Gb3: higher in all subgroups of FD, especially in patients whose disease stage had proceeded. Detection of eight lyso-Gb3-related analogues: lyso-Gb3(-12) and lyso-Gb3(+14) for the first time. Majority of the FD patients had elevated plasma concentrations of the lyso-Gb3 analogues, especially lyso-Gb3(-2). | [116] |
| 55 pediatric Fabry patients, 26 healthy pediatric controls, 108 adult Fabry patients, 16 healthy adult controls | random urine | UPLC-MS/MS | Pediatric FD patients:
| [112] |
| 74 Fabry patients, 41 healthy controls | plasma | UPLC-MS/MS | Lyso-Gb3 and related analogues in plasma:
| [109] |
| 114 Fabry patients, 34 healthy controls | plasma | TOF MS | Fabry patients: three new lyso-Gb3 analogs (m/z ratios at 802, 804, and 820) with higher relative concentration in males compared to female patients. None was detected in the majority in healthy controls. | [102] |
| 24 Fabry patients, 8 healthy controls | plasma | TOF MS | Identification and characterization of five novel analogues/isoforms of Gb3 | [101] |
| 164 Fabry patients, 94 healthy controls | random urine | HPLC-MS/MS | Further analysis of 7 previously discovered lyso-Gb3-related biomarkers:
| [117] |
| 16 male Fabry patients, 16 healthy control males | random urine | TOF MS | 15 isoforms/analogs of Gb3:
| [118] |
| 63 untreated Fabry patients, 59 healthy controls | random urine | TOF MS | Seven novel urinary biomarkers (mass-to-charge ratios (m/z) of 758, 774, 784, 800, 802, 820, and 836)
| [100] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Levstek, T.; Vujkovac, B.; Trebusak Podkrajsek, K. Biomarkers of Fabry Nephropathy: Review and Future Perspective. Genes 2020, 11, 1091. https://doi.org/10.3390/genes11091091
Levstek T, Vujkovac B, Trebusak Podkrajsek K. Biomarkers of Fabry Nephropathy: Review and Future Perspective. Genes. 2020; 11(9):1091. https://doi.org/10.3390/genes11091091
Chicago/Turabian StyleLevstek, Tina, Bojan Vujkovac, and Katarina Trebusak Podkrajsek. 2020. "Biomarkers of Fabry Nephropathy: Review and Future Perspective" Genes 11, no. 9: 1091. https://doi.org/10.3390/genes11091091
APA StyleLevstek, T., Vujkovac, B., & Trebusak Podkrajsek, K. (2020). Biomarkers of Fabry Nephropathy: Review and Future Perspective. Genes, 11(9), 1091. https://doi.org/10.3390/genes11091091