Whole Genome Analysis of Environmental Pseudomonas mendocina Strains: Virulence Mechanisms and Phylogeny



Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacteria Dataset
2.2. Genome Sequencing, Assembly, and Annotation
2.3. Phylogenetic Analysis
2.4. COG Classification, Genomic Islands, and Bacteriophages Prediction
2.5. Antimicrobial Resistance and Virulence Analysis
3. Results
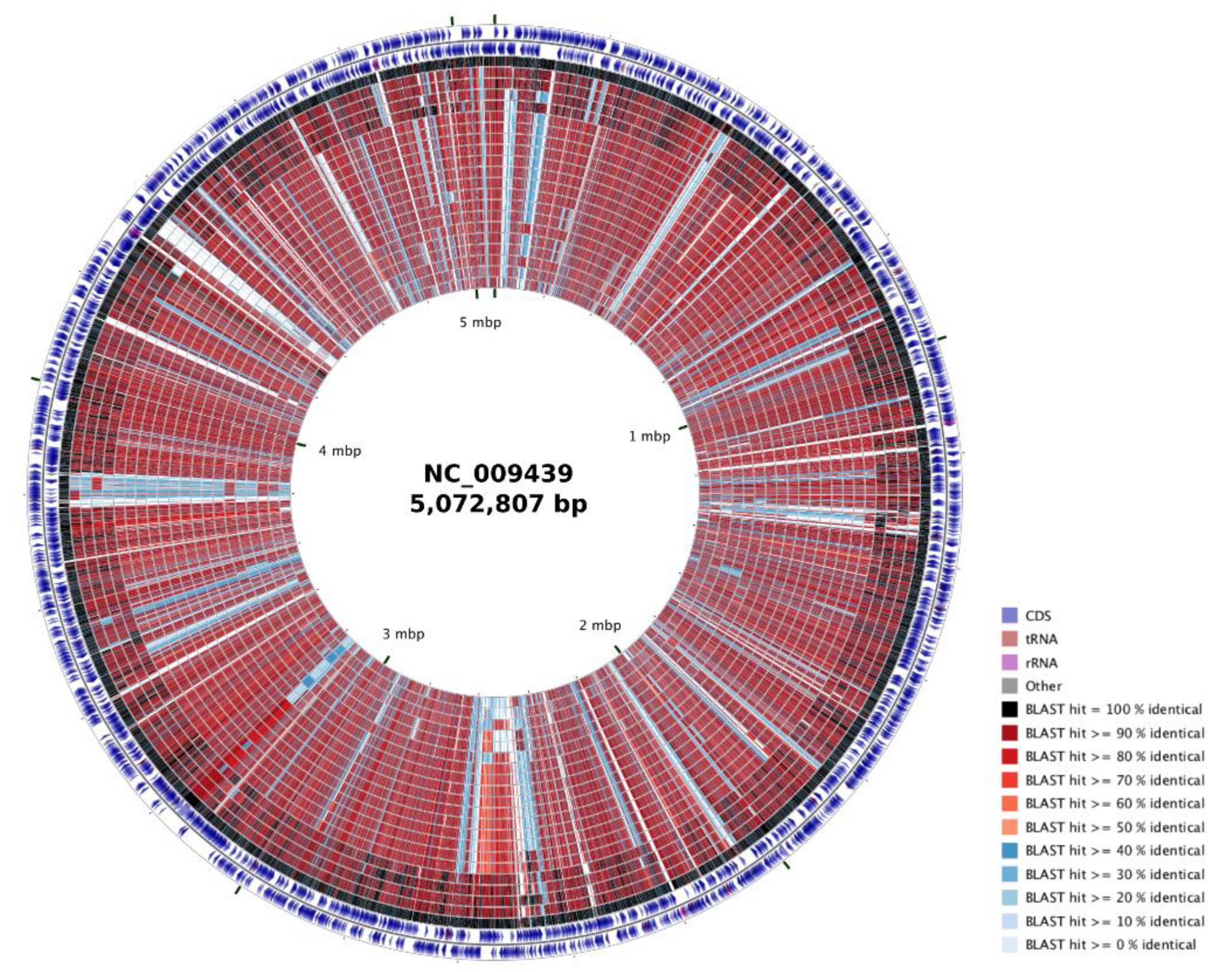
3.1. Genome Properties
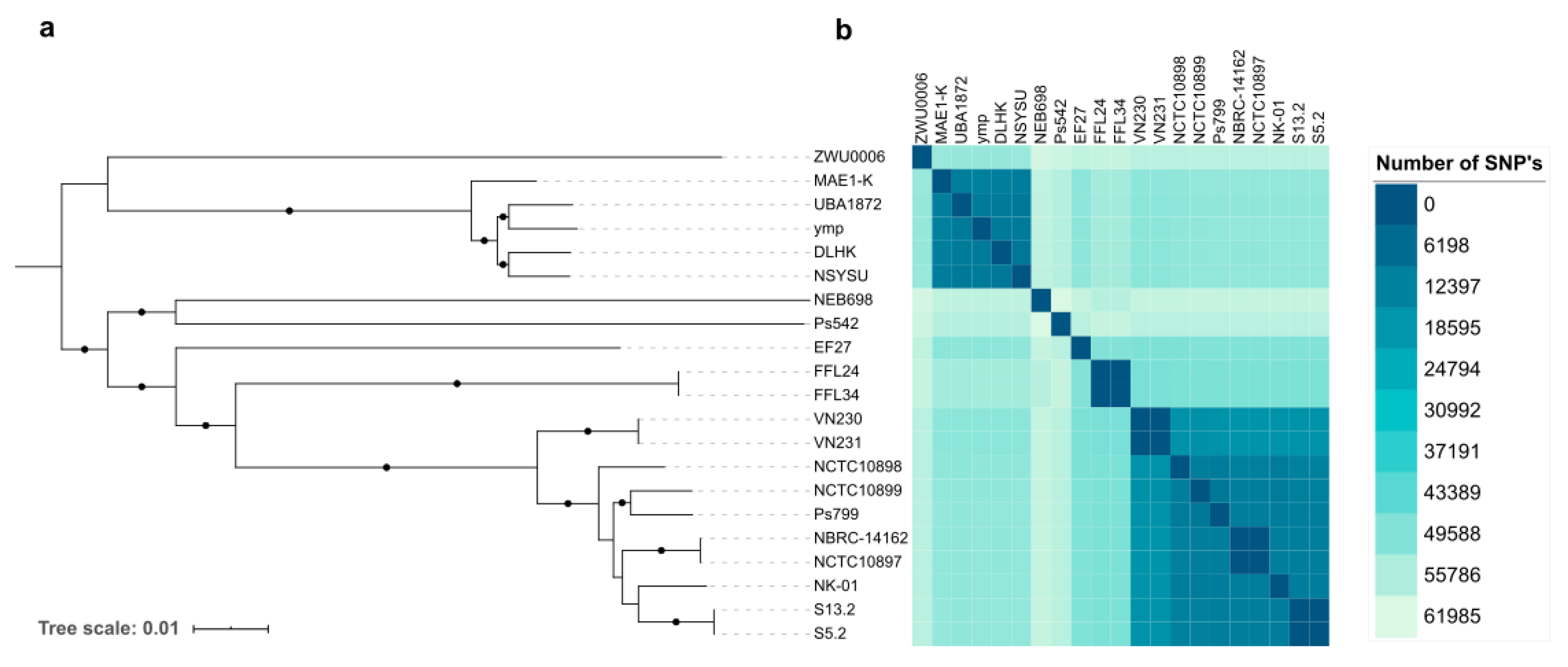
3.2. Phylogenetic Analysis
3.3. COG Classification, Genomic Islands, and Bacteriophages Prediction
3.4. Antimicrobial Resistance and Virulence Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Moradali, M.F.; Ghods, S.; Rehm, B.H.A. Pseudomonas aeruginosa Lifestyle: A Paradigm for Adaptation, Survival, and Persistence. Front. Cell. Infect. Microbiol. 2017, 7, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peix, A.; Ramírez-Bahena, M.-H.; Velázquez, E. Historical evolution and current status of the taxonomy of genus Pseudomonas. Infect. Genet. Evol. 2009, 9, 1132–1147. [Google Scholar] [CrossRef]
- Palleroni, N.J.; Doudoroff, M.; Stanier, R.Y.; Solánes, R.E.; Mandel, M. Taxonomy of the aerobic pseudomonads: The properties of the Pseudomonas stutzeri group. J. Gen. Microbiol. 1970, 60, 215–231. [Google Scholar] [CrossRef] [Green Version]
- Aragone, M.R.; Maurizi, D.M.; Clara, L.O.; Navarro Estrada, J.L.; Ascione, A. Pseudomonas mendocina, an environmental bacterium isolated from a patient with human infective endocarditis. J. Clin. Microbiol. 1992, 30, 1583–1584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nseir, W.; Taha, H.; Abid, A.; Khateeb, J. Pseudomonas mendocina sepsis in a healthy man. Isr. Med. Assoc. J. 2011, 13, 375–376. [Google Scholar] [PubMed]
- Pena, R.T.; Blasco, L.; Ambroa, A.; González-Pedrajo, B.; Fernández-García, L.; López, M.; Bleriot, I.; Bou, G.; García-Contreras, R.; Wood, T.K.; et al. Relationship Between Quorum Sensing and Secretion Systems. Front. Microbiol. 2019, 10, 1100. [Google Scholar] [CrossRef] [Green Version]
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of age: Ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016, 17, 333–351. [Google Scholar] [CrossRef]
- Vernikos, G.; Medini, D.; Riley, D.R.; Tettelin, H. Ten years of pan-genome analyses. Curr. Opin. Microbiol. 2015, 23, 148–154. [Google Scholar] [CrossRef]
- Freschi, L.; Vincent, A.T.; Jeukens, J.; Emond-Rheault, J.-G.; Kukavica-Ibrulj, I.; Dupont, M.-J.; Charette, S.J.; Boyle, B.; Levesque, R.C. The Pseudomonas aeruginosa Pan-Genome Provides New Insights on Its Population Structure, Horizontal Gene Transfer, and Pathogenicity. Genome Biol. Evol. 2019, 11, 109–120. [Google Scholar] [CrossRef] [Green Version]
- Estepa, V.; Rojo-Bezares, B.; Torres, C.; Sáenz, Y. Faecal carriage of Pseudomonas aeruginosa in healthy humans: Antimicrobial susceptibility and global genetic lineages. FEMS Microbiol. Ecol. 2014, 89, 15–19. [Google Scholar] [CrossRef] [Green Version]
- Seppey, M.; Manni, M.; Zdobnov, E.M. BUSCO: Assessing Genome Assembly and Annotation Completeness. In Gene Prediction; Humana: New York, NY, USA, 2019; pp. 227–245. [Google Scholar]
- Vielva, L.; de Toro, M.; Lanza, V.F.; de la Cruz, F. PLACNETw: A web-based tool for plasmid reconstruction from bacterial genomes. Bioinformatics 2017, 33, 3796–3798. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.G.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Grant, J.R.; Arantes, A.S.; Stothard, P. Comparing thousands of circular genomes using the CGView Comparison Tool. BMC Genom. 2012, 13, 202. [Google Scholar] [CrossRef] [Green Version]
- Richter, M.; Rosselló-Móra, R. Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. USA 2009, 106, 19126–19131. [Google Scholar] [CrossRef] [Green Version]
- Goris, J.; Konstantinidis, K.T.; Klappenbach, J.A.; Coenye, T.; Vandamme, P.; Tiedje, J.M. DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 2007, 57, 81–91. [Google Scholar] [CrossRef] [Green Version]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef] [Green Version]
- Team, R.C. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018. [Google Scholar]
- Huerta-Cepas, J.; Forslund, K.; Coelho, L.P.; Szklarczyk, D.; Jensen, L.J.; von Mering, C.; Bork, P. Fast Genome-Wide Functional Annotation through Orthology Assignment by eggNOG-Mapper. Mol. Biol. Evol. 2017, 34, 2115–2122. [Google Scholar] [CrossRef] [Green Version]
- Bertelli, C.; Laird, M.R.; Williams, K.P.; Lau, B.Y.; Hoad, G.; Winsor, G.L.; Brinkman, F.S.L. IslandViewer 4: Expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 2017, 45, W30–W35. [Google Scholar] [CrossRef]
- Mathee, K.; Narasimhan, G.; Valdes, C.; Qiu, X.; Matewish, J.M.; Koehrsen, M.; Rokas, A.; Yandava, C.N.; Engels, R.; Zeng, E.; et al. Dynamics of Pseudomonas aeruginosa genome evolution. Proc. Natl. Acad. Sci. USA. 2008, 105, 3100–3105. [Google Scholar] [CrossRef] [Green Version]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, 16–21. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Liang, Y.; Lynch, K.H.; Dennis, J.J.; Wishart, D.S. PHAST: A Fast Phage Search Tool. Nucleic Acids Res. 2011, 39, 347–352. [Google Scholar] [CrossRef]
- Jurtz, V.I.; Villarroel, J.; Lund, O.; Larsen, M.V.; Nielsen, M. MetaPhinder—Identifying Bacteriophage Sequences in Metagenomic Data Sets. PLoS ONE 2016, 11, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Jia, B.; Raphenya, A.R.; Alcock, B.; Waglechner, N.; Guo, P.; Tsang, K.K.; Lago, B.A.; Dave, B.M.; Pereira, S.; Sharma, A.N.; et al. CARD 2017: Expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 2017, 45, D566–D573. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Zheng, D.; Jin, Q.; Chen, L.; Yang, J. VFDB 2019: A comparative pathogenomic platform with an interactive web interface. Nucleic Acids Res. 2019, 47, D687–D692. [Google Scholar] [CrossRef]
- Emms, D.M.; Kelly, S. OrthoFinder: Solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 2015, 16, 157. [Google Scholar] [CrossRef] [Green Version]
- Alvarado, A.; Garcillán-Barcia, M.P.; de la Cruz, F. A degenerate primer MOB typing (DPMT) method to classify γ-proteobacterial plasmids in clinical and environmental settings. PLoS ONE 2012, 7, e40438. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [Green Version]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Poulsen, B.E.; Yang, R.; Clatworthy, A.E.; White, T.; Osmulski, S.J.; Li, L.; Penaranda, C.; Lander, E.S.; Shoresh, N.; Hung, D.T. Defining the core essential genome of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA. 2019, 116, 10072–10080. [Google Scholar] [CrossRef] [Green Version]
- Freschi, L.; Jeukens, J.; Kukavica-Ibrulj, I.; Boyle, B.; Dupont, M.-J.; Laroche, J.; Larose, S.; Maaroufi, H.; Fothergill, J.L.; Moore, M.; et al. Clinical utilization of genomics data produced by the international Pseudomonas aeruginosa consortium. Front. Microbiol. 2015, 6, 1036. [Google Scholar] [CrossRef] [Green Version]
- Nelson, K.E.; Weinel, C.; Paulsen, I.T.; Dodson, R.J.; Hilbert, H.; Martins dos Santos, V.A.P.; Fouts, D.E.; Gill, S.R.; Pop, M.; Holmes, M.; et al. Complete genome sequence and comparative analysis of the metabolically versatile Pseudomonas putida KT2440. Environ. Microbiol. 2002, 4, 799–808. [Google Scholar] [CrossRef] [Green Version]
- Udaondo, Z.; Molina, L.; Segura, A.; Duque, E.; Ramos, J.L. Analysis of the core genome and pangenome of Pseudomonas putida. Environ. Microbiol. 2016, 18, 3268–3283. [Google Scholar] [CrossRef]
- Jun, S.-R.; Wassenaar, T.M.; Nookaew, I.; Hauser, L.; Wanchai, V.; Land, M.; Timm, C.M.; Lu, T.-Y.S.; Schadt, C.W.; Doktycz, M.J.; et al. Diversity of Pseudomonas Genomes, Including Populus-Associated Isolates, as Revealed by Comparative Genome Analysis. Appl. Environ. Microbiol. 2016, 82, 375–383. [Google Scholar] [CrossRef] [Green Version]
- Freschi, L.; Bertelli, C.; Jeukens, J.; Moore, M.P.; Kukavica-Ibrulj, I.; Emond-Rheault, J.-G.; Hamel, J.; Fothergill, J.L.; Tucker, N.P.; McClean, S.; et al. Genomic characterisation of an international Pseudomonas aeruginosa reference panel indicates that the two major groups draw upon distinct mobile gene pools. FEMS Microbiol. Lett. 2018, 365, fny120. [Google Scholar] [CrossRef] [Green Version]
- Guo, W.; Wang, Y.; Song, C.; Yang, C.; Li, Q.; Li, B.; Su, W.; Sun, X.; Song, D.; Yang, X.; et al. Complete genome of Pseudomonas mendocina NK-01, which synthesizes medium-chain-length polyhydroxyalkanoates and alginate oligosaccharides. J. Bacteriol. 2011, 193, 3413–3414. [Google Scholar] [CrossRef] [Green Version]
- Chong, T.M.; Yin, W.-F.; Mondy, S.; Grandclément, C.; Dessaux, Y.; Chan, K.-G. Heavy-metal resistance of a France vineyard soil bacterium, Pseudomonas mendocina strain S5.2, revealed by whole-genome sequencing. J. Bacteriol. 2012, 194, 6366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, C.F.; Niu, H.; Jiang, J.; Li, J.; Chan, C.M.N.; Leung, D.Y.C.; Leung, F.C.C. Genome Sequence of Pseudomonas mendocina DLHK, Isolated from a Biotrickling Reactor. J. Bacteriol. 2012, 194, 6326. [Google Scholar] [CrossRef] [Green Version]
- Jain, C.; Rodriguez-R, L.M.; Phillippy, A.M.; Konstantinidis, K.T.; Aluru, S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 2018, 9, 5114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzmán-Herrador, D.L.; Llosa, M. The secret life of conjugative relaxases. Plasmid 2019, 104, 102415. [Google Scholar] [CrossRef]
- Garcillán-Barcia, M.P.; Francia, M.V.; de la Cruz, F. The diversity of conjugative relaxases and its application in plasmid classification. FEMS Microbiol. Rev. 2009, 33, 657–687. [Google Scholar] [CrossRef] [Green Version]
- Lanza, V.F.; de Toro, M.; Garcillán-Barcia, M.P.; Mora, A.; Blanco, J.; Coque, T.M.; de la Cruz, F. Plasmid Flux in Escherichia coli ST131 Sublineages, Analyzed by Plasmid Constellation Network (PLACNET), a New Method for Plasmid Reconstruction from Whole Genome Sequences. PLoS Genet. 2014, 10, e1004766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Toro, M.; Fernández, J.; García, V.; Mora, A.; Blanco, J.; de la Cruz, F.; Rodicio, M.R. Whole genome sequencing, molecular typing and in vivo virulence of OXA-48-producing Escherichia coli isolates including ST131 H30-Rx, H22 and H41 subclones. Sci. Rep. 2017, 7, 12103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, X.; Hall, A.B.; Xavier, R.J.; Alm, E.J. Comprehensive analysis of chromosomal mobile genetic elements in the gut microbiome reveals phylum-level niche-adaptive gene pools. PLoS ONE 2019, 14, e0223680. [Google Scholar] [CrossRef] [Green Version]
- Guglielmini, J.; Quintais, L.; Garcillán-Barcia, M.P.; de la Cruz, F.; Rocha, E.P.C. The Repertoire of ICE in Prokaryotes Underscores the Unity, Diversity, and Ubiquity of Conjugation. PLoS Genet. 2011, 7, e1002222. [Google Scholar] [CrossRef] [PubMed]
- Partridge, S.R.; Kwong, S.M.; Firth, N.; Jensen, S.O. Mobile Genetic Elements Associated with Antimicrobial Resistance. Clin. Microbiol. Rev. 2018, 31. [Google Scholar] [CrossRef] [Green Version]
- Rojo-Bezares, B.; Estepa, V.; Cebollada, R.; de Toro, M.; Somalo, S.; Seral, C.; Castillo, F.J.; Torres, C.; Sáenz, Y. Carbapenem-resistant Pseudomonas aeruginosa strains from a Spanish hospital: Characterization of metallo-β-lactamases, porin OprD and integrons. Int. J. Med. Microbiol. 2014, 304, 405–414. [Google Scholar] [CrossRef]
- Almuzara, M.; Montaña, S.; Carulla, M.; Sly, G.; Fernandez, J.; Hernandez, M.; Moriano, A.; Traglia, G.M.; Bakai, R.; Ramirez, M.S. Clinical cases of VIM-producing Pseudomonas mendocina from two burned patients. J. Glob. Antimicrob. Resist. 2018, 14, 273–274. [Google Scholar] [CrossRef]
- Bellés, A.; Bueno, J.; Rojo-Bezares, B.; Torres, C.; Javier Castillo, F.; Sáenz, Y.; Seral, C. Characterisation of VIM-2-producing Pseudomonas aeruginosa isolates from lower tract respiratory infections in a Spanish hospital. Eur. J. Clin. Microbiol. Infect. Dis. 2018, 37, 1847–1856. [Google Scholar] [CrossRef] [PubMed]
- Estepa, V.; Rojo-Bezares, B.; Torres, C.; Sáenz, Y. Genetic Lineages and Antimicrobial Resistance in Pseudomonas spp. Isolates Recovered from Food Samples. Foodborne Pathog. Dis. 2015, 12, 486–491. [Google Scholar] [CrossRef]
- Gani, M.; Rao, S.; Miller, M.; Scoular, S. Pseudomonas mendocina Bacteremia: A Case Study and Review of Literature. Am. J. Case Rep. 2019, 20, 453–458. [Google Scholar] [CrossRef]
- Feldman, M.; Bryan, R.; Rajan, S.; Scheffler, L.; Brunnert, S.; Tang, H.; Prince, A. Role of flagella in pathogenesis of Pseudomonas aeruginosa pulmonary infection. Infect. Immun. 1998, 66, 43–51. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.; Yoon, S.S. Pseudomonas aeruginosa Biofilm, a Programmed Bacterial Life for Fitness. J. Microbiol. Biotechnol. 2017, 27, 1053–1064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nolan, L.M.; Whitchurch, C.B.; Barquist, L.; Katrib, M.; Boinett, C.J.; Mayho, M.; Goulding, D.; Charles, I.G.; Filloux, A.; Parkhill, J.; et al. A global genomic approach uncovers novel components for twitching motility-mediated biofilm expansion in Pseudomonas aeruginosa. Microb. Genom. 2018, 4. [Google Scholar] [CrossRef]
- Giltner, C.L.; Nguyen, Y.; Burrows, L.L. Type IV pilin proteins: Versatile molecular modules. Microbiol. Mol. Biol. Rev. 2012, 76, 740–772. [Google Scholar] [CrossRef] [Green Version]
- Leighton, T.L.; Buensuceso, R.N.C.; Howell, P.L.; Burrows, L.L. Biogenesis of Pseudomonas aeruginosa type IV pili and regulation of their function. Environ. Microbiol. 2015, 17, 4148–4163. [Google Scholar] [CrossRef]
- Valentine, M.E.; Kirby, B.D.; Withers, T.R.; Johnson, S.L.; Long, T.E.; Hao, Y.; Lam, J.S.; Niles, R.M.; Yu, H.D. Generation of a highly attenuated strain of Pseudomonas aeruginosa for commercial production of alginate. Microb. Biotechnol. 2020, 13, 162–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sana, T.G.; Berni, B.; Bleves, S. The T6SSs of Pseudomonas aeruginosa Strain PAO1 and Their Effectors: Beyond Bacterial-Cell Targeting. Front. Cell. Infect. Microbiol. 2016, 6, 61. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Ps542 Genome | Ps799 Genome | |
|---|---|---|
| Assembly parameters | ||
| K-mer size (bp) | 101 | 101 |
| Number of contigs | 64 | 33 |
| Contig maximum length (bp) | 1,348,745 | 2,521,352 |
| N50 (bp) | 467,068 | 734,345 |
| Total bp assembly | 5,187,128 | 5,446,182 |
| Contigs >1 Kb | 22 | 15 |
| Average insert size (bp) | 514 ± 140 | 529 ± 160 |
| Genetic elements | ||
| Size (bp) | 5,178,769 | 5,440,495 |
| GC content (%) | 63.04 | 62.63 |
| Genes | 4725 | 5022 |
| Protein coding genes | 4658 | 4955 |
| Genes with predicted functions | 3198 | 3329 |
| rRNA genes (5S, 16S, 23S) | 1, 1, 1 | 1, 1, 1 |
| tRNA genes | 66 | 66 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruiz-Roldán, L.; de Toro, M.; Sáenz, Y. Whole Genome Analysis of Environmental Pseudomonas mendocina Strains: Virulence Mechanisms and Phylogeny. Genes 2021, 12, 115. https://doi.org/10.3390/genes12010115
Ruiz-Roldán L, de Toro M, Sáenz Y. Whole Genome Analysis of Environmental Pseudomonas mendocina Strains: Virulence Mechanisms and Phylogeny. Genes. 2021; 12(1):115. https://doi.org/10.3390/genes12010115
Chicago/Turabian StyleRuiz-Roldán, Lidia, María de Toro, and Yolanda Sáenz. 2021. "Whole Genome Analysis of Environmental Pseudomonas mendocina Strains: Virulence Mechanisms and Phylogeny" Genes 12, no. 1: 115. https://doi.org/10.3390/genes12010115
APA StyleRuiz-Roldán, L., de Toro, M., & Sáenz, Y. (2021). Whole Genome Analysis of Environmental Pseudomonas mendocina Strains: Virulence Mechanisms and Phylogeny. Genes, 12(1), 115. https://doi.org/10.3390/genes12010115