
Transcriptome Profiling of Maize (Zea mays L.) Leaves Reveals Key Cold-Responsive Genes, Transcription Factors, and Metabolic Pathways Regulating Cold Stress Tolerance at the Seedling Stage

 ,
, 
 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and Treatments
2.2. RNA Extraction, Library Construction, and Illumina Sequencing
2.3. Reads Processing, Mapping, and Gene Expression Quantification
2.4. Functional Annotation of the DEGs
2.5. Gene Ontology (GO) Enrichment and KEGG Pathway Enrichment Analyses
2.6. Validation of Cold-Responsive DEGs by Quantitative Real-Time PCR (qRT-PCR)
3. Results
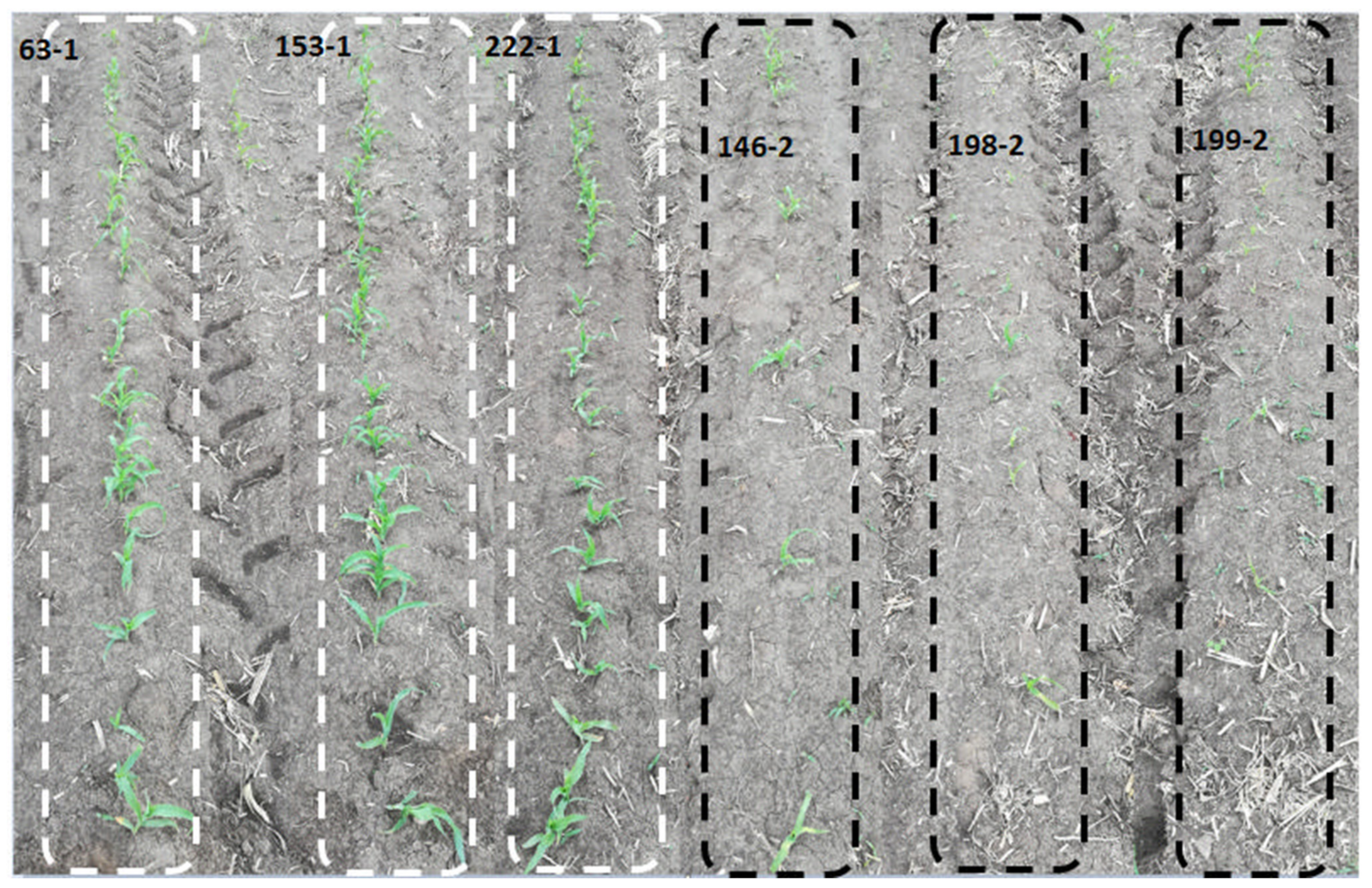
3.1. Phenotypic Analysis of Maize Population under Cold Stress Conditions
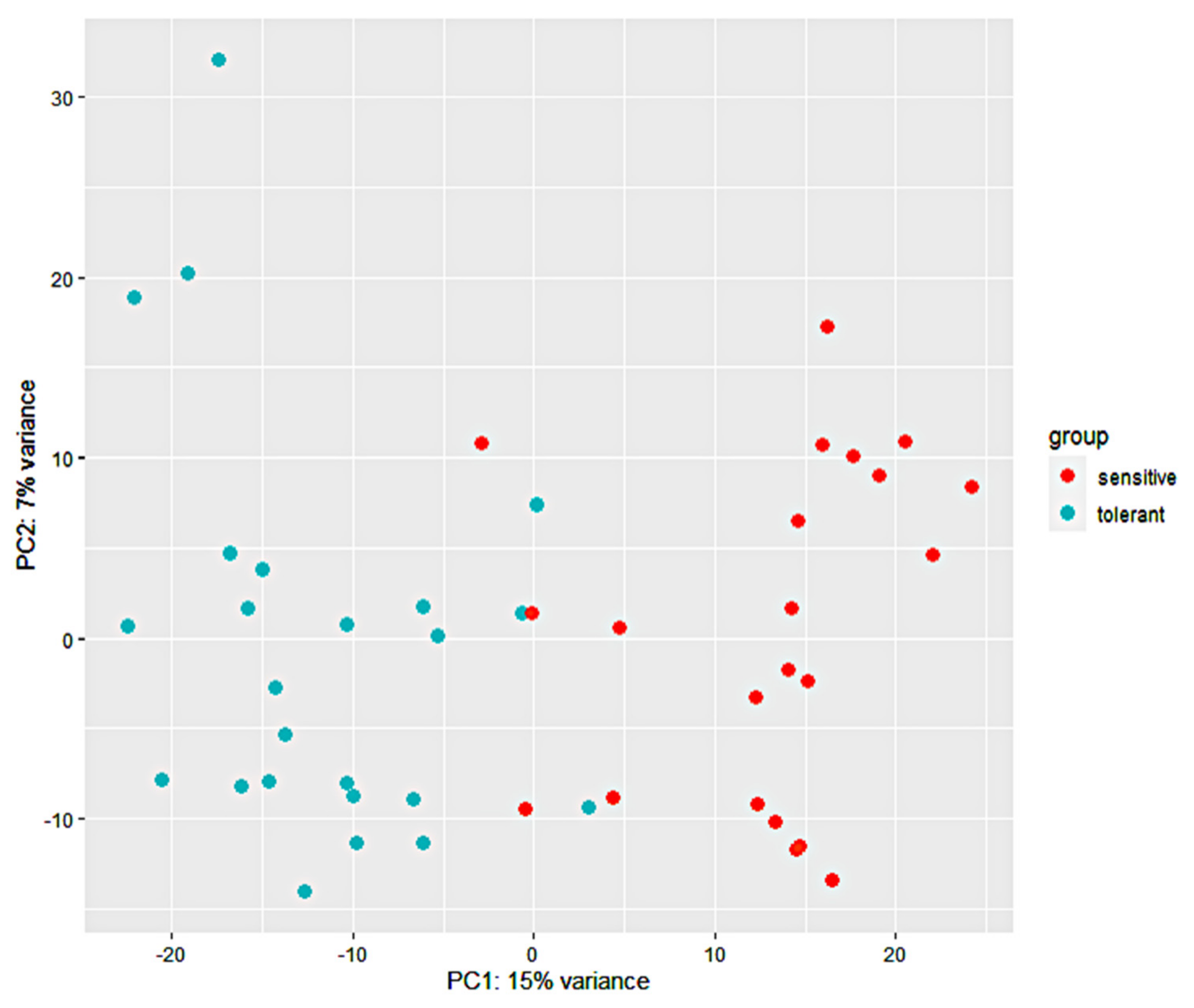
3.2. RNA-Seq Analysis and Alignment of Unique Reads to the Maize Reference Genome
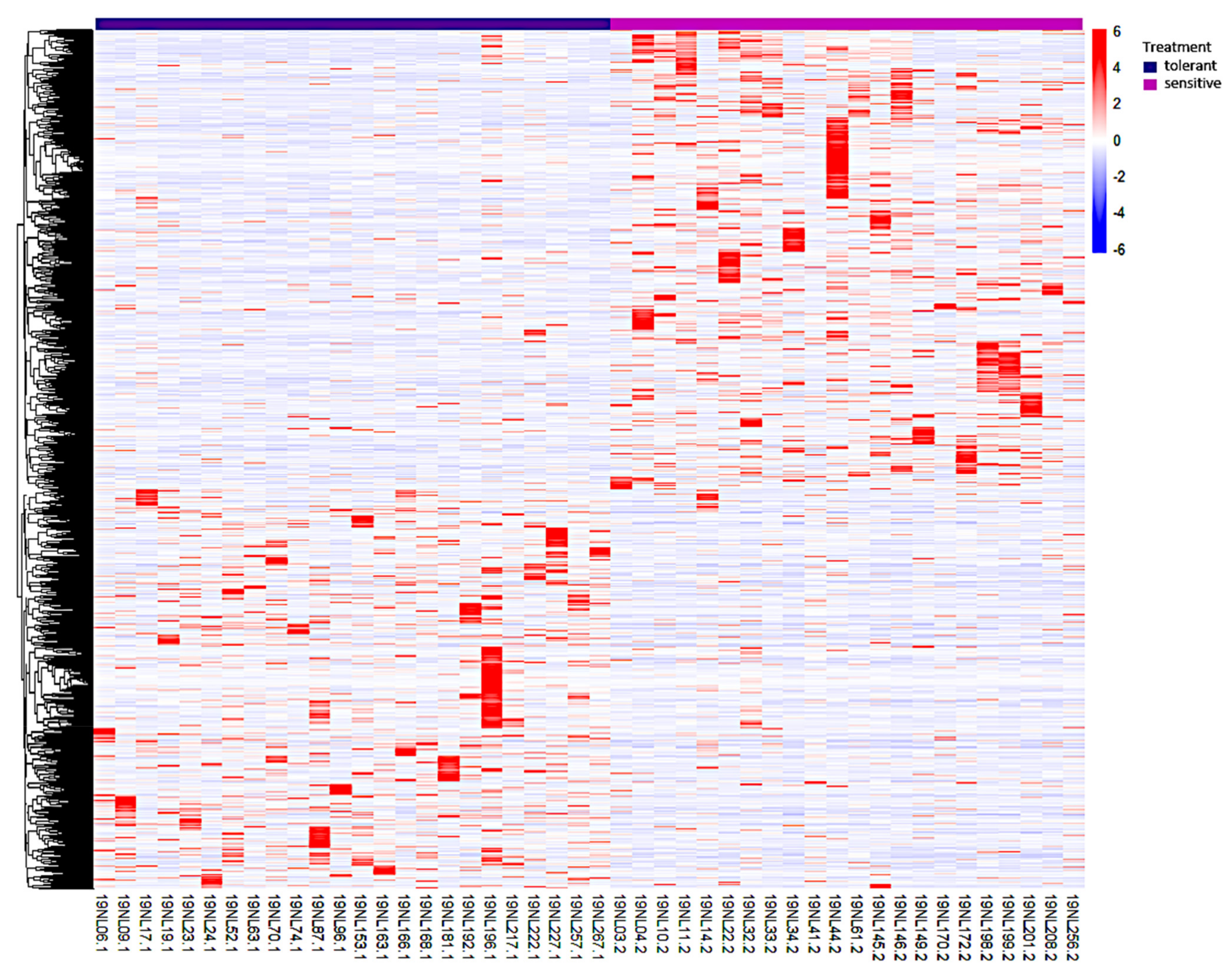
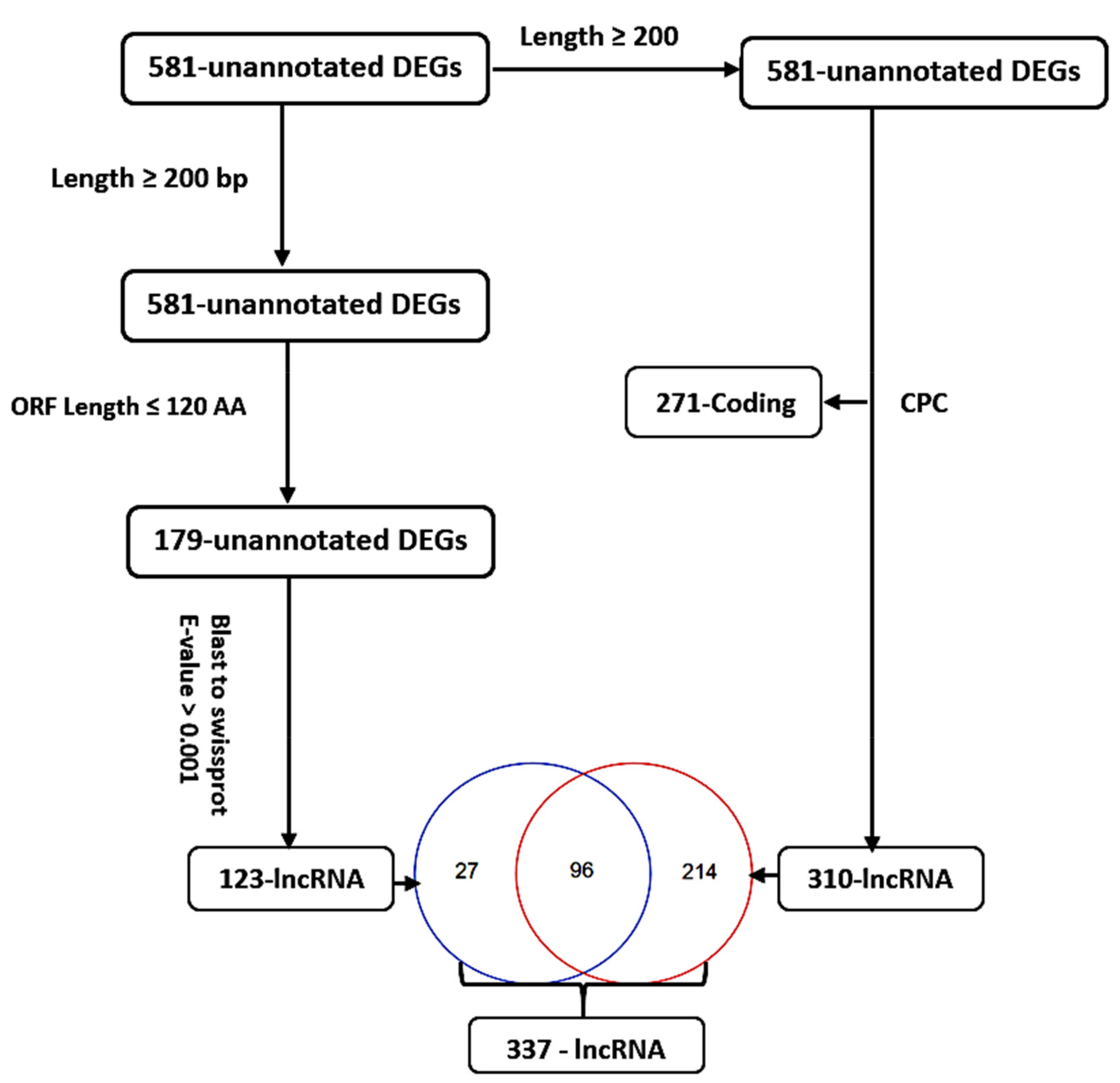
3.3. Identification, Annotation, and Differential Analysis of DEGs
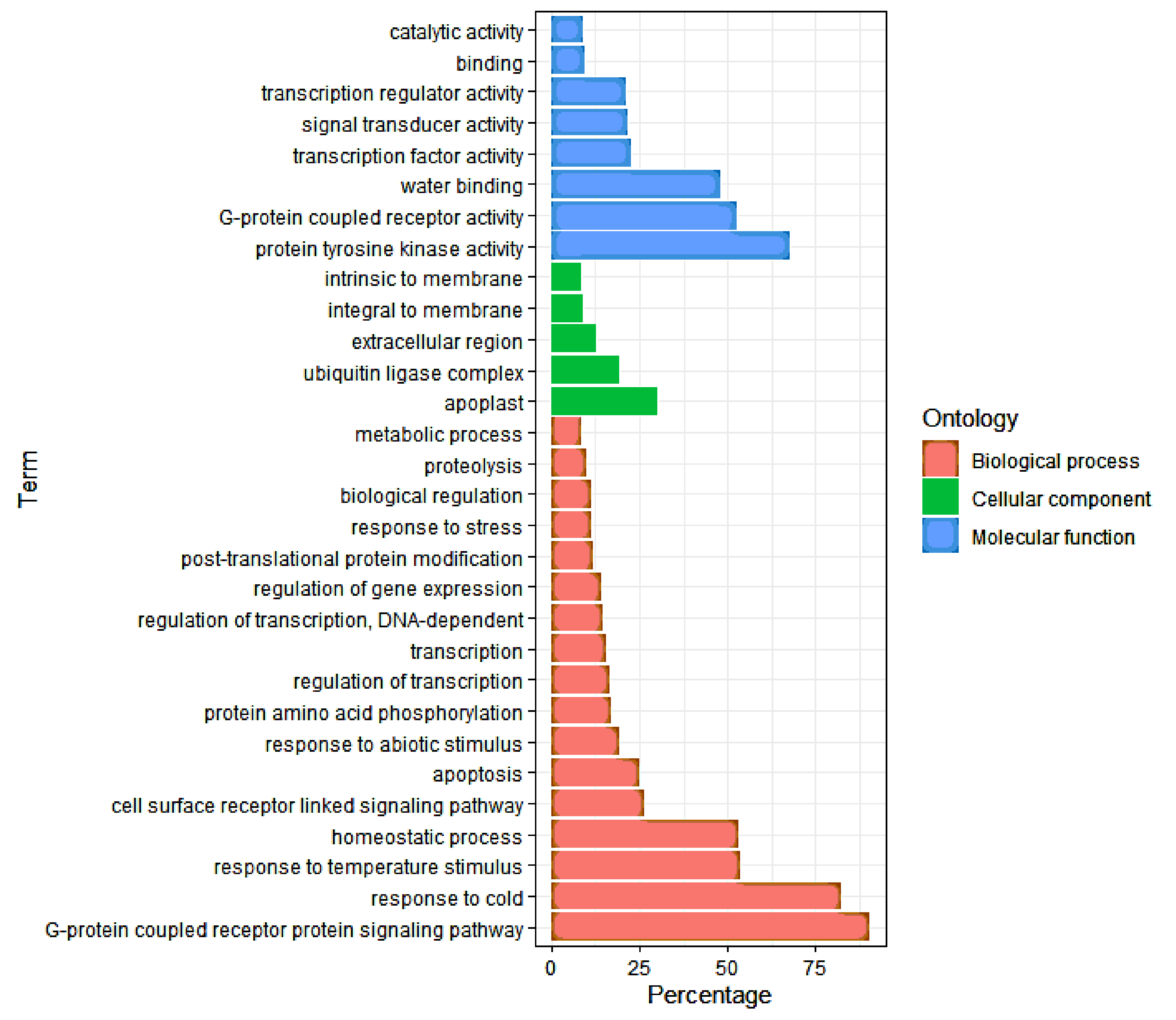
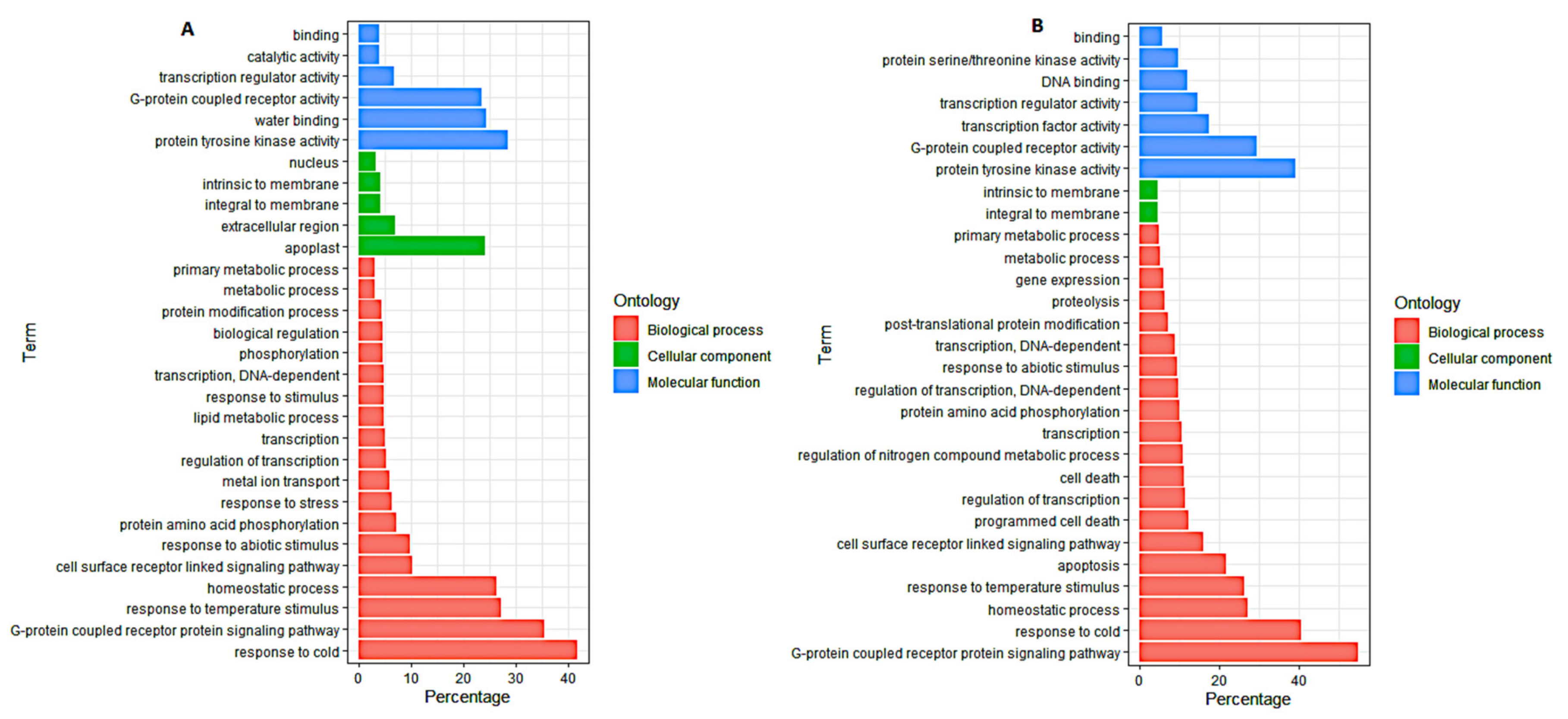
3.4. Gene Ontology (GO) Analysis of DEGs
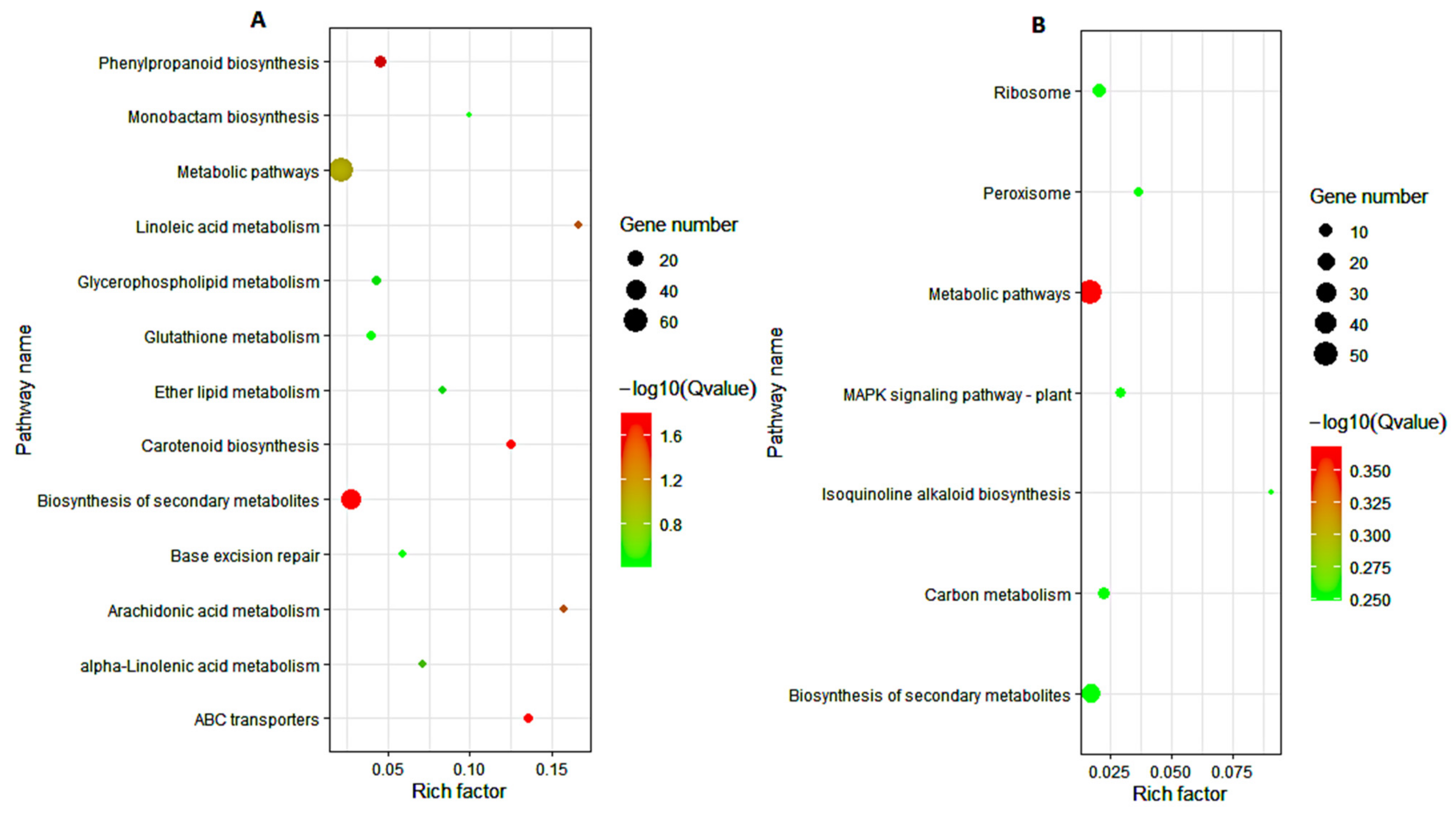
3.5. KEGG Pathway Analysis of DEGs
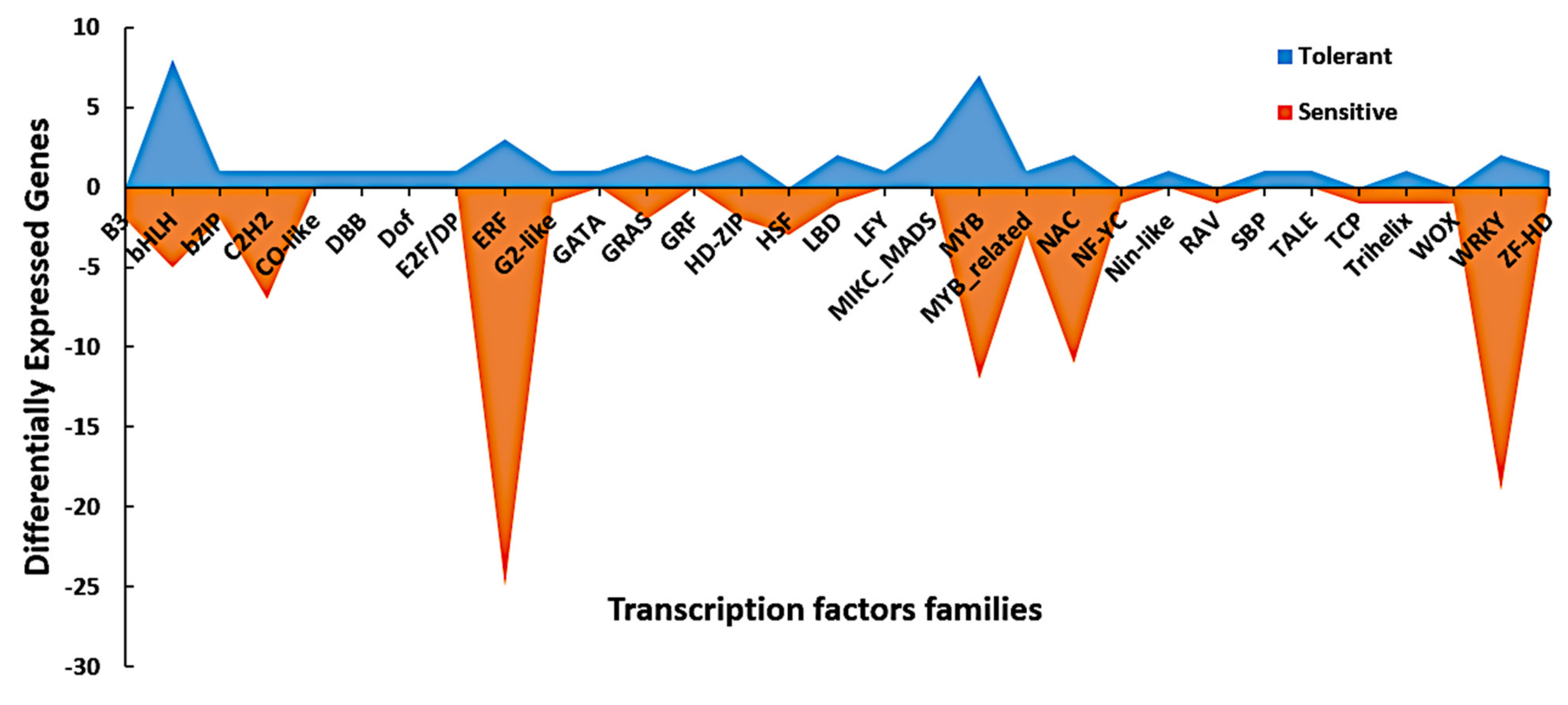
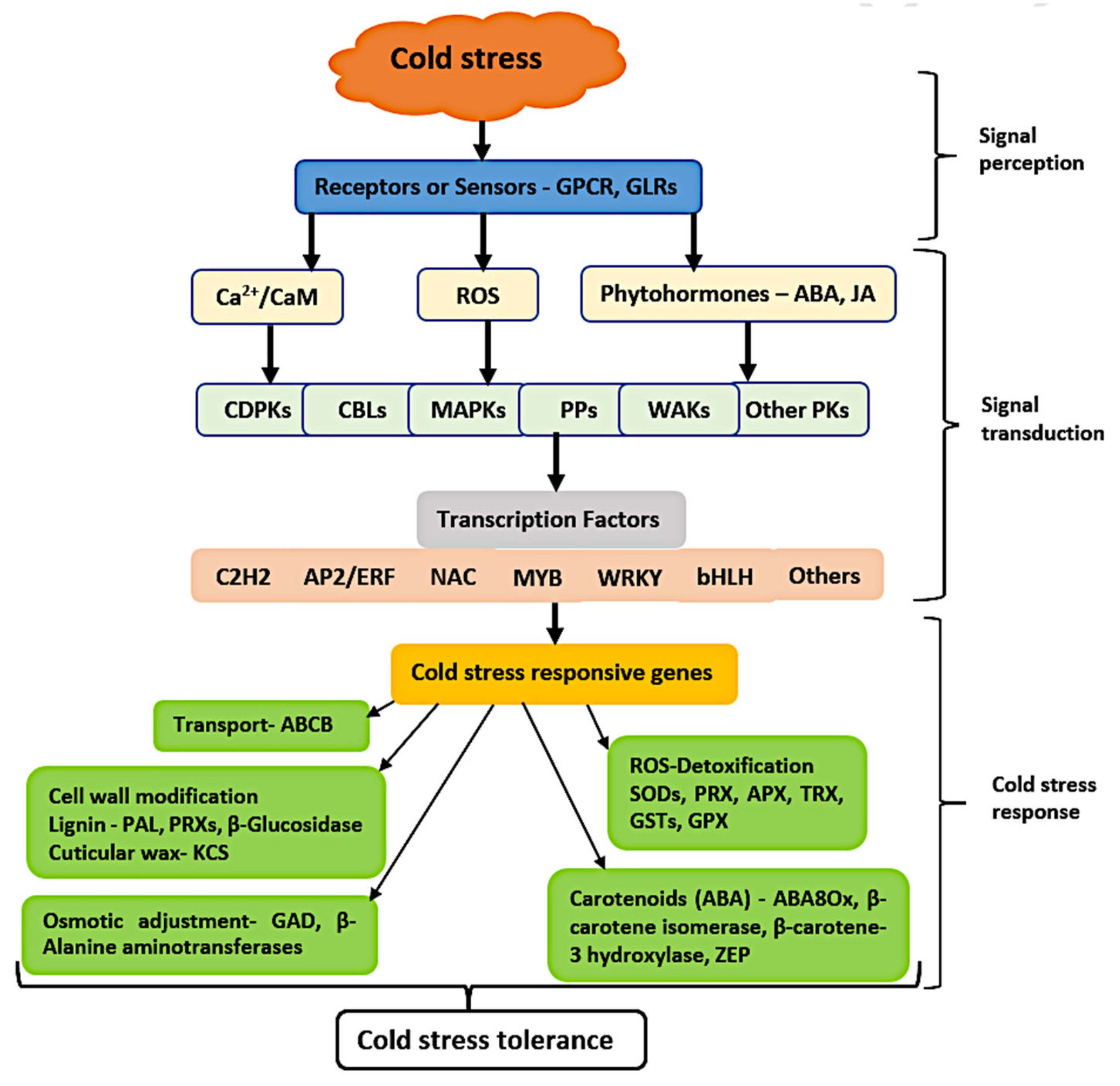
3.6. Dynamic Expression of Signaling and Transcription Factors Genes in Response to Cold Stress
3.7. DEGs Related to “Response to Cold”
3.8. Expression Analysis of Genes Involved in Metabolism, Transport and Functional Impacts of Co-Expressed Gene Hubs
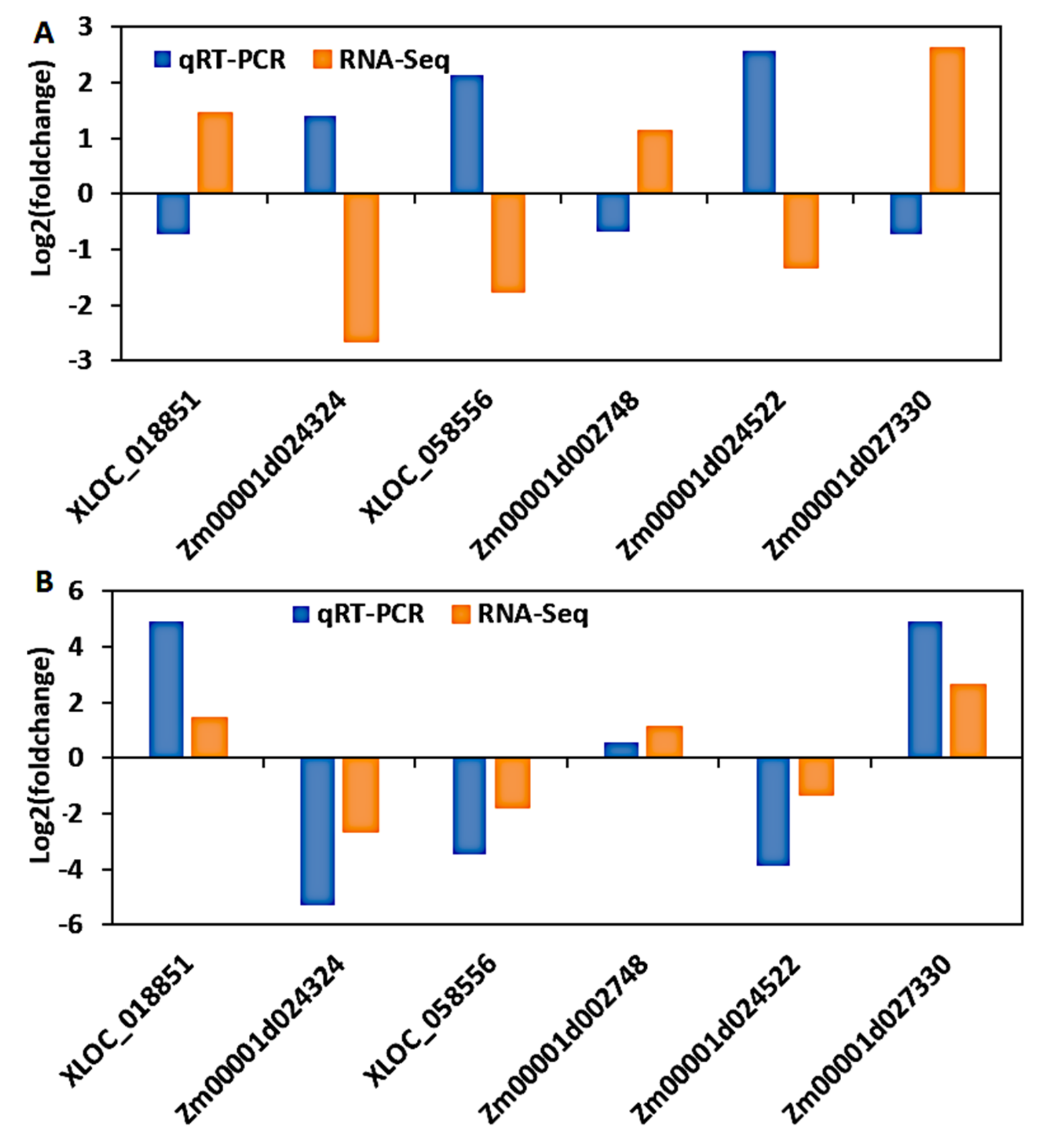
3.9. Validation of DEGs by Quantitative Real-Time PCR (qRT-PCR)
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| bHLH | Basic helix-loop-helix |
| DEGs | Differentially expressed genes |
| MYB | Myeloblastosis |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| GO | Gene Ontology |
| PCA | principal component analysis |
| GPCR | G-protein coupled receptor |
| JA | jasmonic acid |
| PPs | protein phosphatase |
| WAK | wall-associated kinase |
| PAL | phenylalanine ammonia lyase |
| KCS | ketoacyl-CoA synthase |
| GAD | glutamate decarboxylase |
| SOD | Superoxide dismutase |
| APX | Ascorbate peroxidase |
| GLR | Glutamate receptor |
| ROS | Reactive oxygen species |
| ABA | Abscisic acid |
| PK | Protein kinase |
| PRX | Peroxidases |
| CDPK | Calcium dependent protein kinase |
| CaMs | Calmodulin |
| CBLs | Calcineurin B-like |
| CMLs | CaM-related proteins |
| MAPK | Mitogen-activated protein kinase |
References
- United States Department of Agriculture (USDA). World Agricultural Supply and Demand Estimates. 2020. Available online: https://downloads.usda.library.cornell.edu/usda-esmis/files/5q47rn72z/z890sd52n/hd76sk58s/production.pdf (accessed on 12 May 2020).
- Gong, F.L.; Yang, F.; Tai, X.; Wang, W. “Omics” of maize stress response for sustainable food production: Opportunities and challenges. Omics A J. Integr. Biol. 2014, 18, 714–732. [Google Scholar] [CrossRef] [Green Version]
- Stocker, T.F.; Qin, D.; Plattner, G.K.; Tignor, M.M.; Allen, S.K.; Boschung, J.; Nauels, A.; Xia, Y.; Bex, V.; Midgley, P.M. Climate Change 2013: The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of IPCC the Intergovernmental Panel on Climate Change; Cambridge University Press: Cambridge, UK, 2014. [Google Scholar]
- Krasensky, J.; Jonak, C. Drought, salt, and temperature stress-induced metabolic rearrangements and regulatory networks. J. Exp. Bot. 2012, 63, 1593–1608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peleg, Z.; Blumwald, E. Hormone balance and abiotic stress tolerance in crop plants. Curr. Opin. Plant Biol. 2011, 14, 290–295. [Google Scholar] [CrossRef]
- Ma, S.Q.; Xi, Z.X.; Wang, Q. Risk evaluation of cold damage to corn in Northeast China. J. Nat. Disasters 2003, 12, 137–141. [Google Scholar]
- Greaves, J.A. Improving suboptimal temperature tolerance in maize-the search for variation. J. Exp. Bot. 1996, 47, 307–323. [Google Scholar] [CrossRef]
- Marocco, A.; Lorenzoni, C.; Fracheboud, Y. Chilling stress in maize. Maydica 2005, 50, 571–580. [Google Scholar]
- Sobkowiak, A.; Jończyk, M.; Adamczyk, J.; Szczepanik, J.; Solecka, D.; Kuciara, I.; Hetmańczyk, K.; Trzcinska-Danielewicz, T.; Grzybowski, M.; Skoneczny, M. Molecular foundations of chilling-tolerance of modern maize. BMC Genom. 2016, 17, 125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leipner, J.; Stamp, P. Chilling Stress in Maize Seedlings. In Handbook of Maize: Its Biology; Springer: New York, NY, USA, 2009; pp. 291–310. [Google Scholar]
- Sowiński, P.; Rudzińska-Langwald, A.; Dalbiak, A.; Sowińska, A. Assimilate export from leaves of chilling-treated seedlings of maize. The path to vein. Plant Physiol. Biochem. 2001, 39, 881–889. [Google Scholar] [CrossRef]
- Zhang, B.; Yang, L.; Li, Y. Comparison of physiological and biochemical characteristics related to cold resistance in sugarcane under field conditions. Acta Agron. Sin. 2011, 37, 496–505. [Google Scholar] [CrossRef]
- Virdi, A.S.; Singh, S.; Singh, P. Abiotic stress responses in plants: Roles of calmodulin-regulated proteins. Front. Plant Sci. 2015, 6, 809. [Google Scholar] [CrossRef] [Green Version]
- Chinnusamy, V.; Zhu, J.; Zhu, J.K. Cold stress regulation of gene expression in plants. Trends Plant Sci. 2007, 12, 444–451. [Google Scholar] [CrossRef] [PubMed]
- Chinnusamy, V.; Zhu, J.K.; Sunkar, R. Gene Regulation During Cold Stress Acclimation in Plants. In Plant Stress Tolerance; Springer: Totowa, NJ, USA, 2010; Volume 639, pp. 39–55. [Google Scholar]
- Zhao, C.; Wang, P.; Si, T.; Hsu, C.C.; Wang, L.; Zayed, O.; Yu, Z.; Zhu, Y.; Dong, J.; Tao, W.A. MAP kinase cascades regulate the cold response by modulating ICE1 protein stability. Dev. Cell 2017, 43, 618–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Ding, Y.; Yang, S. Cold signal transduction and its interplay with phytohormones during cold acclimation. Plant Cell Physiol. 2015, 56, 7–15. [Google Scholar] [CrossRef]
- Sah, S.K.; Reddy, K.R.; Li, J. Abscisic acid and abiotic stress tolerance in crop plants. Front. Plant Sci. 2016, 7, 571. [Google Scholar] [CrossRef] [Green Version]
- Tarkowski, Ł.P.; Van den Ende, W. Cold tolerance triggered by soluble sugars: A multifaceted countermeasure. Front. Plant Sci. 2015, 6, 203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnholdt-Schmitt, B.; Costa, J.H.; de Melo, D.F. AOX—A functional marker for efficient cell reprogramming under stress? Trends Plant Sci. 2006, 11, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Al-Whaibi, M.H. Plant heat-shock proteins: A mini review. J. King Saud Univ. Sci. 2011, 23, 139–150. [Google Scholar] [CrossRef] [Green Version]
- Alejandro, S.; Lee, Y.; Tohge, T.; Sudre, D.; Osorio, S.; Park, J.; Bovet, L.; Lee, Y.; Geldner, N.; Fernie, A.R. AtABCG29 is a monolignol transporter involved in lignin biosynthesis. Curr. Biol. 2012, 22, 1207–1212. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Guo, G.; Wang, C.; Lin, Y.; Wang, X.; Zhao, M.; Guo, Y.; He, M.; Zhang, Y.; Pan, L. Survey of the transcriptome of Aspergillus oryzae via massively parallel mRNA sequencing. Nucleic Acids Res. 2010, 38, 5075–5087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Huang, R. Enhanced tolerance to freezing in tobacco and tomato overexpressing transcription factor TERF2/LeERF2 is modulated by ethylene biosynthesis. Plant Mol. Biol. 2010, 73, 241–249. [Google Scholar] [CrossRef]
- Jończyk, M.; Sobkowiak, A.; Trzcinska-Danielewicz, J.; Sowiński, P. Chromatin-Level Differences Elucidate Potential Determinants of Contrasting Levels of Cold Sensitivity in Maize Lines. Plant Mol. Biol. Report. 2021, 39, 335–350. [Google Scholar] [CrossRef]
- Grzybowski, M.; Adamczyk, J.; Jończyk, M.; Sobkowiak, A.; Szczepanik, J.; Frankiewicz, K.; Sowiński, P. Increased photosensitivity at early growth as a possible mechanism of maize adaptation to cold springs. J. Exp. Bot. 2019, 70, 2887–2904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, A.; Pimentel, H.; Trapnell, C.; Pachter, L. Identification of novel transcripts in annotated genomes using RNA-Seq. Bioinformatics 2011, 27, 2325–2329. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Nat. Preced. 2010. [Google Scholar] [CrossRef]
- Li, L.; Eichten, S.R.; Shimizu, R.; Petsch, K.; Yeh, C.T.; Wu, W.; Chettoor, A.M.; Givan, S.A.; Cole, R.A.; Fowler, J.E. Genome-wide discovery and characterization of maize long non-coding RNAs. Genome Biol. 2014, 15, R40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, L.; Zhang, Y.; Ye, Z.Q.; Liu, X.Q.; Zhao, S.Q.; Wei, L.; Gao, G. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007, 35, W345–W349. [Google Scholar] [CrossRef]
- Tian, T.; Liu, Y.; Yan, H.; You, Q.; Yi, X.; Du, Z.; Xu, W.; Su, Z. agriGO v2. 0: A GO analysis toolkit for the agricultural community, 2017 update. Nucleic Acids Res. 2017, 45, W122–W129. [Google Scholar] [CrossRef]
- Supek, F.; Bošnjak, M.; Škunca, N.; Šmuc, T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.Y.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Zhu, L.; Xue, J.; Yang, J.; Hu, H.; Cui, J.; Xu, J. Selection and Verification of Appropriate Reference Genes for Expression Normalization in Cryptomeria fortunei Under Abiotic Stress and Hormone Treatments. Genes 2021, 12, 791. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Novaković, L.; Guo, T.; Bacic, A.; Sampathkumar, A.; Johnson, K.L. Hitting the wall—Sensing and signaling pathways involved in plant cell wall remodeling in response to abiotic stress. Plants 2018, 7, 89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, J.; Tian, F.; Yang, D.C.; Meng, Y.Q.; Kong, L.; Luo, J.; Gao, G. PlantTFDB 4.0: Toward a central hub for transcription factors and regulatory interactions in plants. Nucleic Acids Res. 2016, 45, gkw982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruz, R.P.D.; Sperotto, R.A.; Cargnelutti, D.; Adamski, J.M.; de FreitasTerra, T.; Fett, J.P. Avoiding damage and achieving cold tolerance in rice plants. Food Energy Secur. 2013, 2, 96–119. [Google Scholar] [CrossRef]
- Li, Y.; Wang, X.; Li, Y.; Zhang, Y.; Gou, Z.; Qi, X.; Zhang, J. Transcriptomic Analysis Revealed the Common and Divergent Responses of Maize Seedling Leaves to Cold and Heat Stresses. Genes 2020, 11, 881. [Google Scholar] [CrossRef]
- Ji, C.Y.; Kim, H.S.; Lee, C.J.; Kim, S.E.; Lee, H.U.; Nam, S.S.; Kwak, S.S. Comparative transcriptome profiling of tuberous roots of two sweet potato lines with contrasting low temperature tolerance during storage. Gene 2020, 727, 144244. [Google Scholar] [CrossRef]
- Yu, T.; Zhang, J.; Cao, J.; Cai, Q.; Li, X.; Sun, Y.; Duan, Y. Leaf transcriptomic response mediated by cold stress in two maize inbred lines with contrasting tolerance levels. Genomics 2021, 113, 782–794. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.K. Abiotic stress signaling and responses in plants. Cell 2016, 167, 313–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, L.; Liu, Y.; Mu, Y.; Anwar, A.; He, C.; Yan, Y.; Li, Y.; Yu, X. Heterotrimeric G-protein γ subunit CsGG3. 2 positively regulates the expression of CBF genes and chilling tolerance in cucumber. Front. Plant Sci. 2018, 9, 488. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Liu, D.; Chong, K. Cold signaling in plants: Insights into mechanisms and regulation. J. Integr. Plant Biol. 2018, 60, 745–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xi, J.; Qiu, Y.; Du, L.; Poovaiah, B.W. Plant-specific trihelix transcription factor AtGT2L interacts with calcium/calmodulin and responds to cold and salt stresses. Plant Sci. 2012, 185, 274–280. [Google Scholar] [CrossRef]
- Xie, Z.M.; Zou, H.F.; Lei, G.; Wei, W.; Zhou, Q.Y.; Niu, C.F.; Chen, S.Y. Soybean Trihelix transcription factors GmGT-2A and GmGT-2B improve plant tolerance to abiotic stresses in transgenic Arabidopsis. PLoS ONE 2009, 4, e6898. [Google Scholar] [CrossRef] [Green Version]
- Sanders, D.; Pelloux, J.; Brownlee, C.; Harper, J.F. Calcium at the crossroads of signaling. Plant Cell 2002, 14, S401–S417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luan, S.; Kudla, J.; Rodriguez-Concepcion, M.; Yalovsky, S.; Gruissem, W. Calmodulins and calcineurin B–like proteins: Calcium sensors for specific signal response coupling in plants. Plant Cell 2002, 14, S389–S400. [Google Scholar] [CrossRef] [Green Version]
- Pareek, A.; Khurana, A.; Sharma, K.; Kumar, R. An overview of signaling regulons during cold stress tolerance in plants. Curr. Genom. 2017, 18, 498–511. [Google Scholar] [CrossRef]
- Li, Q.; Lei, S.; Du, K.; Li, L.; Pang, X.; Wang, Z.; Xu, L. RNA-seq based transcriptomic analysis uncovers α-linolenic acid and jasmonic acid biosynthesis pathways respond to cold acclimation in Camellia japonica. Sci. Rep. 2016, 6, 36463. [Google Scholar] [CrossRef]
- Wu, X.; Bacic, A.; Johnson, K.L.; Humphries, J. The role of brachypodium distachyon wall-associated kinases (WAKs) in cell expansion and stress responses. Cells 2020, 9, 2478. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.G.; Waadt, R.; Cheong, Y.H.; Pandey, G.K.; Dominguez-Solis, J.R.; Schültke, S.; Lee, S.C.; Kudla, J.; Luan, S. The calcium sensor CBL10 mediates salt tolerance by regulating ion homeostasis in Arabidopsis. Plant J. 2007, 52, 473–484. [Google Scholar] [CrossRef]
- Lee, B.H.; Henderson, D.A.; Zhu, J.K. The Arabidopsis cold-responsive transcriptome and its regulation by ICE1. Plant Cell 2005, 17, 3155–3175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez, F.A.P.; Estrada, Y.; Flores, F.B.; Ortíz-Atienza, A.; Lozano, R.; Egea, I. The Ca2+ Sensor Calcineurin B-like Protein 10 in Plants: Emerging New Crucial Roles for Plant Abiotic Stress Tolerance. Front. Plant Sci. 2020, 11, 2155. [Google Scholar]
- Kamal, M.M.; Ishikawa, S.; Takahashi, F.; Suzuki, K.; Kamo, M.; Umezawa, T.; Uemura, M. Large-Scale Phosphoproteomic Study of Arabidopsis Membrane Proteins Reveals Early Signaling Events in Response to Cold. Int. J. Mol. Sci. 2020, 21, 8631. [Google Scholar] [CrossRef]
- Kumar, M.; Gho, Y.S.; Jung, K.H.; Kim, S.R. Genome-wide identification and analysis of genes, conserved between japonica and indica rice cultivars that respond to low-temperature stress at the vegetative growth stage. Front. Plant Sci. 2017, 8, 1120. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; He, Y.; Zhu, Y.; Li, M.; Song, S.; Bo, W.; Pang, X. Comparative transcriptome profiling reveals cold stress responsiveness in two contrasting Chinese jujube cultivars. BMC Plant Biol. 2020, 20, 240. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, Y.; He, Y.; Hu, W.; Zhang, Y.; Wang, X.; Tang, H. Identification of NADPH oxidase family members associated with cold stress in strawberry. FEBS Open Bio 2018, 8, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Zhang, L.; Wang, H.; Lu, J.; Wei, H.; Yu, S. Transcriptomic Profiling of Young Cotyledons Response to Chilling Stress in Two Contrasting Cotton (Gossypium hirsutum L.) Genotypes at the Seedling Stage. Int. J. Mol. Sci. 2020, 21, 5095. [Google Scholar] [CrossRef] [PubMed]
- Kovtun, Y.; Chiu, W.L.; Tena, G.; Sheen, J. Functional analysis of oxidative stress-activated mitogen-activated protein kinase cascade in plants. Proc. Natl. Acad. Sci. USA 2000, 97, 2940–2945. [Google Scholar] [CrossRef] [Green Version]
- Schweighofer, A.; Hirt, H.; Meskiene, I. Plant PP2C phosphatases: Emerging functions in stress signaling. Trends Plant Sci. 2004, 9, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Hu, X.; Song, J.; Zong, X.; Li, D.; Li, D. Over-expression of a Zea mays L. protein phosphatase 2C gene (ZmPP2C) in Arabidopsis thaliana decreases tolerance to salt and drought. J. Plant Physiol. 2009, 166, 531–542. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.G.; Seo, P.J. The MYB 96–HHP module integrates cold and abscisic acid signaling to activate the CBF–COR pathway in Arabidopsis. Plant J. 2015, 82, 962–977. [Google Scholar] [CrossRef] [PubMed]
- Meng, J.; Hu, B.; Yi, G.; Li, X.; Chen, H.; Wang, Y.; Xu, C. Genome-wide analyses of banana fasciclin-like AGP genes and their differential expression under low-temperature stress in chilling sensitive and tolerant cultivars. Plant Cell Rep. 2020, 39, 693–708. [Google Scholar] [CrossRef] [PubMed]
- Martín, M.L.; Busconi, L. A rice membrane-bound calcium-dependent protein kinase is activated in response to low temperature. Plant Physiol. 2001, 125, 1442–1449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwal, M.; Hao, Y.; Kapoor, A.; Dong, C.H.; Fujii, H.; Zheng, X.; Zhu, J.K. A R2R3 type MYB transcription factor is involved in the cold regulation of CBF genes and in acquired freezing tolerance. J. Biol. Chem. 2006, 281, 37636–37645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pradhan, S.K.; Pandit, E.; Nayak, D.K.; Behera, L.; Mohapatra, T. Genes, pathways and transcription factors involved in seedling stage chilling stress tolerance in indica rice through RNA-Seq analysis. BMC Plant Biol. 2019, 19, 352. [Google Scholar] [CrossRef] [Green Version]
- Jiang, C.; Zhang, H.; Ren, J.; Dong, J.; Zhao, X.; Wang, X.; Yu, H. Comparative Transcriptome-Based Mining and Expression Profiling of Transcription Factors Related to Cold Tolerance in Peanut. Int. J. Mol. Sci. 2020, 21, 1921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Övernäs, E.; Sundås Larsson, A.; Söderman, E. Two AP2 Transcription Factors, AtERF38 and AtERF39, Have Similar Function but are Differently Regulated in ABA and Stress Responses; Uppsala University Publications: Upsala, Sweden, 2010. [Google Scholar]
- Krishnaswamy, S.; Verma, S.; Rahman, M.H.; Kav, N.N. Functional characterization of four APETALA2-family genes (RAP2. 6, RAP2. 6L, DREB19 and DREB26) in Arabidopsis. Plant Mol. Biol. 2011, 75, 107–127. [Google Scholar] [CrossRef]
- Wang, J.; Tao, F.; An, F.; Zou, Y.; Tian, W.; Chen, X.; Xu, X.; Hu, X. Wheat transcription factor TaWRKY70 is positively involved in high-temperature seedling plant resistance to Puccinia striiformis f. sp. tritici. Mol. Plant Pathol. 2017, 18, 649–661. [Google Scholar] [CrossRef]
- Wei, W.; Zhang, Y.; Han, L.; Guan, Z.; Chai, T. A novel WRKY transcriptional factor from Thlaspi caerulescens negatively regulates the osmotic stress tolerance of transgenic tobacco. Plant Cell Rep. 2008, 27, 795–803. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Chen, X.; Dong, S.; Jiang, X.; Wang, L.; Yu, J.; Zhou, Y. Crosstalk of PIF4 and DELLA modulates CBF transcript and hormone homeostasis in cold response in tomato. Plant Biotechnol. J. 2020, 18, 1041–1055. [Google Scholar] [CrossRef]
- Li, Z.; Liu, C.; Zhang, Y.; Wang, B.; Ran, Q.; Zhang, J. The bHLH family member ZmPTF1 regulates drought tolerance in maize by promoting root development and abscisic acid synthesis. J. Exp. Bot. 2019, 70, 5471–5486. [Google Scholar] [CrossRef] [PubMed]
- Vannini, C.; Locatelli, F.; Bracale, M.; Magnani, E.; Marsoni, M.; Osnato, M.; Mattana, M.; Baldoni, E.; Coraggio, I. Overexpression of the rice Osmyb4 gene increases chilling and freezing tolerance of Arabidopsis thaliana plants. Plant J. 2004, 37, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Babitha, K.; Vemanna, R.S.; Nataraja, K.N.; Udayakumar, M. Overexpression of EcbHLH57 transcription factor from Eleusine coracana L. in tobacco confers tolerance to salt, oxidative and drought stress. PLoS ONE 2015, 10, e0137098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khatun, K.; Robin, A.H.K.; Park, J.I.; Nath, U.K.; Kim, C.K.; Lim, K.B.; Nou, I.S.; Chung, M.Y. Molecular characterization and expression profiling of tomato GRF transcription factor family genes in response to abiotic stresses and phytohormones. Int. J. Mol. Sci. 2017, 18, 1056. [Google Scholar] [CrossRef] [Green Version]
- Vogel, J.T.; Zarka, D.G.; Van Buskirk, H.A.; Fowler, S.G.; Thomashow, M.F. Roles of the CBF2 and ZAT12 transcription factors in configuring the low temperature transcriptome of Arabidopsis. Plant J. 2005, 41, 195–211. [Google Scholar] [CrossRef]
- Tang, S.; Xian, Y.; Wang, F.; Luo, C.; Song, W.; Xie, S.; Liu, H. Comparative transcriptome analysis of leaves during early stages of chilling stress in two different chilling-tolerant brown-fiber cotton cultivars. PLoS ONE 2021, 16, e0246801. [Google Scholar] [CrossRef]
- Agarwal, P.K.; Jha, B. Transcription factors in plants and ABA dependent and independent abiotic stress signaling. Biol. Plant 2010, 54, 201–212. [Google Scholar] [CrossRef]
- Chen, J.; Tian, Q.; Pang, T.; Jiang, L.; Wu, R.; Xia, X.; Yin, W. Deep-sequencing transcriptome analysis of low temperature perception in a desert tree, Populus euphratica. BMC Genom. 2014, 15, 326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baron, K.N.; Schroeder, D.F.; Stasolla, C. Transcriptional response of abscisic acid (ABA) metabolism and transport to cold and heat stress applied at the reproductive stage of development in Arabidopsis thaliana. Plant Sci. 2012, 188, 48–59. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Wang, Y.; Chang, L.; Zhang, T.; An, J.; Liu, Y.; Cao, Y.; Zhao, X.; Sha, X.; Hu, T. MsZEP, a novel zeaxanthin epoxidase gene from alfalfa (Medicago sativa), confers drought and salt tolerance in transgenic tobacco. Plant Cell Rep. 2016, 35, 439–453. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Wang, N.; Cui, F.; Li, X.; Xiao, J.; Xiong, L. Characterization of the β-carotene hydroxylase gene DSM2 conferring drought and oxidative stress resistance by increasing xanthophylls and abscisic acid synthesis in rice. Plant Physiol. 2010, 154, 1304–1318. [Google Scholar] [CrossRef] [Green Version]
- Nayyar, H.; Chander, S. Protective effects of polyamines against oxidative stress induced by water and cold stress in chickpea. J. Agron. Crop Sci. 2004, 190, 355–365. [Google Scholar] [CrossRef]
- Li, Y.; Wang, X.; Ban, Q.; Zhu, X.; Jiang, C.; Wei, C.; Bennetzen, J.L. Comparative transcriptomic analysis reveals gene expression associated with cold adaptation in the tea plant Camellia sinensis. BMC Genom. 2019, 20, 624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.; Li, C.X.; Zhong, J.; Shu, D.; Luo, D.; Li, Z.M.; Ma, X.R. Exogenous 1′, 4′-trans-Diol-ABA Induces Stress Tolerance by Affecting the Level of Gene Expression in Tobacco (Nicotiana tabacum L.). Int. J. Mol. Sci. 2021, 22, 2555. [Google Scholar] [CrossRef] [PubMed]
- Filiz, E.; Ozyigit, I.I.; Saracoglu, I.A.; Uras, M.E.; Sen, U.; Yalcin, B. Abiotic stress-induced regulation of antioxidant genes in different Arabidopsis ecotypes: Microarray data evaluation. Biotechnol. Biotechnol. Equip. 2019, 33, 128–143. [Google Scholar] [CrossRef] [Green Version]
- Baek, K.H.; Skinner, D.Z. Alteration of antioxidant enzyme gene expression during cold acclimation of near-isogenic wheat lines. Plant Sci. 2003, 165, 1221–1227. [Google Scholar] [CrossRef]
- Rezaie, R.; Mandoulakani, B.A.; Fattahi, M. Cold stress changes antioxidant defense system, phenylpropanoid contents and expression of genes involved in their biosynthesis in Ocimum basilicum L. Sci. Rep. 2020, 10, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Ma, J.; Zhang, Q.; Wu, C.; Zhao, H.; Wu, Y.; He, G. Genome-wide identification and expression profiling of glutathione transferase gene family under multiple stresses and hormone treatments in wheat (Triticum aestivum L.). BMC Genom. 2019, 20, 986. [Google Scholar] [CrossRef] [Green Version]
- Takesawa, T.; Ito, M.; Kanzaki, H.; Kameya, N.; Nakamura, I. Overexpression of ζ glutathione S-transferase in transgenic rice enhances germination and growth at low temperature. Mol. Breed. 2002, 9, 93–101. [Google Scholar] [CrossRef]
- Xu, J.; Yang, J.; Duan, X.; Jiang, Y.; Zhang, P. Increased expression of native cytosolic Cu/Zn superoxide dismutase and ascorbate peroxidase improves tolerance to oxidative and chilling stresses in cassava (Manihot esculenta Crantz). BMC Plant Biol. 2014, 14, 208. [Google Scholar] [CrossRef] [PubMed]
- Almagro, L.; Gómez Ros, L.V.; Belchi-Navarro, S.; Bru, R.; Ros Barceló, A.; Pedreno, M.A. Class III peroxidases in plant defence reactions. J. Exp. Bot. 2009, 60, 377–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, Y.; Siala, W.; Bashandy, T.; Riondet, C.; Vignols, F.; Reichheld, J.P. Glutaredoxins and thioredoxins in plants. Biochim. Biophys. Acta Mol. Cell Res. 2008, 1783, 589–600. [Google Scholar] [CrossRef]
- Ortiz, D.; Hu, J.; Salas Fernandez, M.G. Genetic architecture of photosynthesis in Sorghum bicolor under non-stress and cold stress conditions. J. Exp. Bot. 2017, 68, 4545–4557. [Google Scholar] [CrossRef] [PubMed]
- Khaledian, Y.; Maali-Amiri, R.; Talei, A. Phenylpropanoid and antioxidant changes in chickpea plants during cold stress. Russ. J. Plant Physiol. 2015, 62, 772–778. [Google Scholar] [CrossRef]
- Srivastava, S.; Vishwakarma, R.K.; Arafat, Y.A.; Gupta, S.K.; Khan, B.M. Abiotic stress induces change in Cinnamoyl CoA Reductase (CCR) protein abundance and lignin deposition in developing seedlings of Leucaena leucocephala. Physiol. Mol. Biol. Plants 2015, 21, 197–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chun-yan, F. Research progress on 4-coumarate: Coenzyme A ligase (4CL) of plants. Mod. Agric. Sci. Technol. 2010, 8, 39–40. [Google Scholar]
- Pandey, V.; Awasthi, M.; Singh, S.; Tiwari, S.; Dwivedi, U. A comprehensive review on function and application of plant peroxidases. Biochem. Anal. Biochem. 2017, 6, 308. [Google Scholar] [CrossRef]
- Naikoo, M.I.; Dar, M.I.; Raghib, M.F.; Jaleel, H.; Ahmad, B.; Raina, A.; Khan, F.A.; Naushin, F. Role and regulation of plants phenolics in abiotic stress tolerance: An overview. Plant Signal. Mol. 2019, 157–168. [Google Scholar] [CrossRef]
- Vogt, T. Phenylpropanoid biosynthesis. Mol. Plant 2010, 3, 2–20. [Google Scholar] [CrossRef] [Green Version]
- Dharmawardhana, D.P.; Ellis, B.E.; Carlson, J.E. A [β]-Glucosidase from lodge pole pine xylem specific for the lignin precursor coniferin. Plant Physiol. 1995, 107, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Kleczkowski, K.; Schell, J.; Bandur, R. Phytohormones conjugates: Nature and function. Crit. Rev. Plant. Sci. 1995, 14, 283–298. [Google Scholar] [CrossRef]
- Baba, S.A.; Vishwakarma, R.A.; Ashraf, N. Functional characterization of CsBGlu12, a β-glucosidase from Crocus sativus, provides insights into its role in abiotic stress through accumulation of antioxidant flavonols. J. Biol. Chem. 2017, 292, 4700–4713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slama, I.; Abdelly, C.; Bouchereau, A.; Flowers, T.; Savoure, A. Diversity, distribution and roles of osmoprotective compounds accumulated in halophytes under abiotic stress. Ann. Bot. 2015, 115, 433–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanson, A.D.; Rathinasabapathi, B.; Chamberlin, B.; Gage, D.A. Comparative physiological evidence that β-alanine betaine and choline-O-sulfate act as compatible osmolytes in halophytic Limonium species. Plant Physiol. 1991, 97, 1199–1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rocha, M.; Licausi, F.; Araujo, W.L.; Nunes-Nesi, A.; Sodek, L.; Fernie, A.R.; van Dongen, J.T. Glycolysis and the tricarboxylic acid cycle are linked by alanine aminotransferase during hypoxia induced by waterlogging of Lotus japonicus. Plant Physiol. 2010, 152, 1501–1513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renault, H.; Roussel, V.; El Amrani, A.; Arzel, M.; Renault, D.; Bouchereau, A.; Deleu, C. The Arabidopsis pop2-1 mutant reveals the involvement of GABA transaminase in salt stress tolerance. BMC Plant Biol. 2010, 10, 20. [Google Scholar] [CrossRef] [Green Version]
- Mazzucotelli, E.; Tartari, A.; Cattivelli, L.; Forlani, G. Metabolism of γ-aminobutyric acid during cold acclimation and freezing and its relationship to frost tolerance in barley and wheat. J. Exp. Bot. 2006, 57, 3755–3766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Dong, J.I.; Wang, J. Research progress in membrane lipid metabolism and molecular mechanism in peanut cold tolerance. Front. Plant Sci. 2019, 10, 838. [Google Scholar] [CrossRef]
- Hu, Y.; Jiang, Y.; Han, X.; Wang, H.; Pan, J.; Yu, D. Jasmonate regulates leaf senescence and tolerance to cold stress: Crosstalk with other phytohormones. J. Exp. Bot. 2017, 68, 1361–1369. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Jiang, L.; Wang, F.; Yu, D. Jasmonate regulates the inducer of CBF expression–c-repeat binding factor/DRE binding factor1 cascade and freezing tolerance in Arabidopsis. Plant Cell 2013, 25, 2907–2924. [Google Scholar] [CrossRef] [Green Version]
- Vergnolle, C.; Vaultier, M.N.; Taconnat, L.; Renou, J.P.; Kader, J.C.; Zachowski, A.; Ruelland, E. The cold-induced early activation of phospholipase C and D pathways determines the response of two distinct clusters of genes in Arabidopsis cell suspensions. Plant Physiol. 2005, 139, 1217–1233. [Google Scholar] [CrossRef] [Green Version]
- Gustavsson, M.H.; Sommarin, M. Characterisation of a plasma membrane-associated phospholipase A2 activity increased in response to cold acclimation. J. Plant Physiol. 2002, 159, 1219–1227. [Google Scholar] [CrossRef]
- Craig, W.; Lenzi, P.; Scotti, N.; De Palma, M.; Saggese, P.; Carbone, V.; Curran, N.M.; Magee, A.M.; Medgyesy, P.; Kavanagh, T.A. Transplastomic tobacco plants expressing a fatty acid desaturase gene exhibit altered fatty acid profiles and improved cold tolerance. Transgenic Res. 2008, 17, 769–782. [Google Scholar] [CrossRef] [PubMed]
- Bernard, A.; Joubès, J. Arabidopsis Cuticular waxes: Advances in synthesis, export and regulation. Prog. Lipid Res. 2013, 52, 110–129. [Google Scholar] [CrossRef]
- Hong, J.K.; Choi, H.W.; Hwang, I.S.; Kim, D.S.; Kim, N.H.; Choi, D.S.; Kim, Y.J.; Hwang, B.K. Function of a novel GDSL-type pepper lipase gene, CaGLIP1, in disease susceptibility and abiotic stress tolerance. Planta 2008, 227, 539–558. [Google Scholar] [CrossRef]
- Hincha, D.K.; Neukamm, B.; Sror, H.A.; Sieg, F.; Weckwarth, W.; Rückels, M.; Schmitt, J.M. Cabbage cryoprotectin is a member of the nonspecific plant lipid transfer protein gene family. Plant Physiol. 2001, 125, 835–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, K.; Zhong, X. Non-specific lipid transfer proteins in maize. BMC Plant Biol. 2014, 14, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Osakabe, Y.; Yamaguchi-Shinozaki, K.; Shinozaki, K.; Tran, L.S.P. ABA control of plant macroelement membrane transport systems in response to water deficit and high salinity. New Phytol. 2014, 202, 35–49. [Google Scholar] [CrossRef]
- Schroeder, J.I.; Delhaize, E.; Frommer, W.B.; Guerinot, M.L.; Harrison, M.J.; Herrera-Estrella, L.; Sanders, D. Using membrane transporters to improve crops for sustainable food production. Nature 2013, 497, 60–66. [Google Scholar] [CrossRef] [Green Version]
- Seki, M.; Narusaka, M.; Ishida, J.; Nanjo, T.; Fujita, M.; Oono, Y.; Shinozaki, K. Monitoring the expression profiles of 7000 Arabidopsis genes under drought, cold and high-salinity stresses using a full-length cDNA microarray. Plant J. 2002, 31, 279–292. [Google Scholar] [CrossRef]
- Chai, C.; Subudhi, P.K. Comprehensive analysis and expression profiling of the OsLAX and OsABCB auxin transporter gene families in rice (Oryza sativa) under phytohormone stimuli and abiotic stresses. Front. Plant Sci. 2016, 7, 593. [Google Scholar] [CrossRef] [Green Version]
- Yue, R.; Tie, S.; Sun, T.; Zhang, L.; Yang, Y.; Qi, J.; Yan, S.; Han, X.; Wang, H.; Shen, C. Genome-wide identification and expression profiling analysis of ZmPIN, ZmPILS, ZmLAX and ZmABCB auxin transporter gene families in maize (Zea mays L.) under various abiotic stresses. PLoS ONE 2015, 10, e0118751. [Google Scholar] [CrossRef] [PubMed]
- Noiraud, N.; Maurousset, L.; Lemoine, R. Transport of polyols in higher plants. Plant Physiol. Biochem. 2001, 39, 717–728. [Google Scholar] [CrossRef]
- Lu, P.; Magwanga, R.O.; Kirungu, J.N.; Hu, Y.; Dong, Q.; Cai, X.; Liu, F. Overexpression of cotton a DTX/MATE gene enhances drought, salt, and cold stress tolerance in transgenic Arabidopsis. Front. Plant Sci. 2019, 10, 299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di, C.; Yuan, J.; Wu, Y.; Li, J.; Lin, H.; Hu, L.; Zhang, T.; Qi, Y.; Gerstein, M.B.; Guo, Y. Characterization of stress-responsive lncRNAs in Arabidopsis thaliana by integrating expression, epigenetic and structural features. Plant J. 2014, 80, 848–861. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Yu, X.; Lei, N.; Cheng, Z.; Zhao, P.; He, Y.; Wang, W.; Peng, M. Genome-wide identification and functional prediction of cold and/or drought-responsive lncRNAs in cassava. Sci. Rep. 2017, 7, 45981. [Google Scholar] [CrossRef]
- Wang, P.; Dai, L.; Ai, J.; Wang, Y.; Ren, F. Identification and functional prediction of cold-related long non-coding RNA (lncRNA) in grapevine. Sci. Rep. 2019, 9, 6638. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TF Family | DEGs in the Tolerant Line | DEGs in the Sensitive Line | Total |
|---|---|---|---|
| B3 | 0 | 2 | 2 |
| GRF | 1 | 0 | 1 |
| ERF | 3 | 25 | 28 |
| DBB | 1 | 0 | 1 |
| Dof | 1 | 0 | 1 |
| HSF | 0 | 3 | 3 |
| LBD | 2 | 1 | 3 |
| LFY | 1 | 0 | 1 |
| MYB | 7 | 12 | 19 |
| NAC | 2 | 11 | 13 |
| RAV | 0 | 1 | 1 |
| SBP | 1 | 0 | 1 |
| WOX | 0 | 1 | 1 |
| TCP | 0 | 1 | 1 |
| E2F/DP | 1 | 0 | 1 |
| GATA | 1 | 0 | 1 |
| GRAS | 2 | 2 | 4 |
| bZIP | 1 | 2 | 3 |
| C2H2 | 1 | 7 | 8 |
| bHLH | 8 | 5 | 13 |
| Nin-like | 1 | 0 | 1 |
| NF-YC | 0 | 1 | 1 |
| ZF-HD | 1 | 0 | 1 |
| G2-like | 1 | 1 | 2 |
| CO-like | 1 | 0 | 1 |
| HD-ZIP | 2 | 2 | 4 |
| WRKY | 2 | 19 | 21 |
| TALE | 1 | 0 | 1 |
| Trihelix | 1 | 1 | 2 |
| MIKC_MADS | 3 | 0 | 3 |
| MYB_related | 1 | 3 | 4 |
| Total | 47 | 100 | 147 |
| Locus ID | Gene ID | log2 Fold Change (T/S) | p_Value | Chr | Start | End | Annotation |
|---|---|---|---|---|---|---|---|
| XLOC_000983 | Zm00001d027606 | 23.73473512 | 6.81 × 10−31 | Chr1 | 8,915,719 | 8,918,057 | transmembrane protein |
| XLOC_056725 | - | −23.7343301 | 9.38 × 10−29 | Chr7 | 170,162,017 | 170,164,579 | - |
| XLOC_046285 | Zm00001d017622 | −2.670016539 | 9.74 × 10−25 | Chr5 | 201,997,628 | 201,998,691 | OSJNBa0088A01 |
| XLOC_018249 | - | 24.01063195 | 1.05 × 10−24 | Chr2 | 137,103,173 | 137,104,433 | - |
| XLOC_058213 | Zm00001d021006 | −1.554984072 | 3.70 × 10−22 | Chr7 | 140,181,294 | 140,183,018 | MTD1 |
| XLOC_034912 | - | 23.0239833 | 5.49 × 10−21 | Chr4 | 176,582,912 | 176,584,805 | - |
| XLOC_056365 | Zm00001d021394 | 22.79156651 | 7.11 × 10−21 | Chr7 | 150,891,109 | 150,895,395 | hypothetical protein |
| XLOC_067464 | - | 20.05873411 | 6.49 × 10−20 | Chr9 | 20,669,455 | 20,671,879 | - |
| XLOC_049980 | - | 24.72008389 | 2.60 × 10−17 | Chr6 | 114,599,197 | 114,601,220 | - |
| XLOC_018351 | - | 24.4739764 | 5.35 × 10−17 | Chr2 | 150,920,460 | 150,923,282 | - |
| XLOC_055724 | - | 22.96632545 | 9.06 × 10−17 | Chr7 | 95,897,276 | 95,897,694 | - |
| XLOC_018159 | Zm00001d004620 | 22.6740887 | 1.68 × 10−16 | Chr2 | 122,442,335 | 122,447,585 | uncharacterized protein |
| XLOC_004771 | - | −23.94951263 | 2.29 × 10−16 | Chr1 | 1,487,983 | 1,491,882 | - |
| XLOC_007853 | Zm00001d033411 | −23.88106934 | 2.77 × 10−16 | Chr1 | 262,279,726 | 262,300,105 | hypothetical protein |
| XLOC_005508 | Zm00001d028673 | 23.87942745 | 2.96 × 10−16 | Chr1 | 42,581,898 | 42,586,482 | small nuclear protein G |
| XLOC_036989 | - | −23.84216727 | 3.11 × 10−16 | Chr4 | 108,230,941 | 108,236,238 | - |
| XLOC_014865 | Zm00001d025968 | 23.6089741 | 6.37 × 10−16 | Chr1 | 134,994,735 | 134,998,645 | hypothetical protein |
| XLOC_018011 | Zm00001d004338 | 23.58304546 | 6.85 × 10−16 | Chr2 | 104,459,685 | 104,460,441 | hypothetical protein |
| XLOC_046112 | Zm00001d017287 | −4.317481518 | 2.92 × 10−13 | Chr5 | 191,750,113 | 191,750,625 | uncharacterized protein |
| XLOC_027574 | Zm00001d043525 | −1.350259753 | 4.09 × 10−13 | Chr3 | 202,741,852 | 202,743,157 | oxidative stress 3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Waititu, J.K.; Cai, Q.; Sun, Y.; Sun, Y.; Li, C.; Zhang, C.; Liu, J.; Wang, H. Transcriptome Profiling of Maize (Zea mays L.) Leaves Reveals Key Cold-Responsive Genes, Transcription Factors, and Metabolic Pathways Regulating Cold Stress Tolerance at the Seedling Stage. Genes 2021, 12, 1638. https://doi.org/10.3390/genes12101638
Waititu JK, Cai Q, Sun Y, Sun Y, Li C, Zhang C, Liu J, Wang H. Transcriptome Profiling of Maize (Zea mays L.) Leaves Reveals Key Cold-Responsive Genes, Transcription Factors, and Metabolic Pathways Regulating Cold Stress Tolerance at the Seedling Stage. Genes. 2021; 12(10):1638. https://doi.org/10.3390/genes12101638
Chicago/Turabian StyleWaititu, Joram Kiriga, Quan Cai, Ying Sun, Yinglu Sun, Congcong Li, Chunyi Zhang, Jun Liu, and Huan Wang. 2021. "Transcriptome Profiling of Maize (Zea mays L.) Leaves Reveals Key Cold-Responsive Genes, Transcription Factors, and Metabolic Pathways Regulating Cold Stress Tolerance at the Seedling Stage" Genes 12, no. 10: 1638. https://doi.org/10.3390/genes12101638
APA StyleWaititu, J. K., Cai, Q., Sun, Y., Sun, Y., Li, C., Zhang, C., Liu, J., & Wang, H. (2021). Transcriptome Profiling of Maize (Zea mays L.) Leaves Reveals Key Cold-Responsive Genes, Transcription Factors, and Metabolic Pathways Regulating Cold Stress Tolerance at the Seedling Stage. Genes, 12(10), 1638. https://doi.org/10.3390/genes12101638