
An Atypical Case of Congenital Erythropoietic Porphyria

 , , and
, , and
Abstract
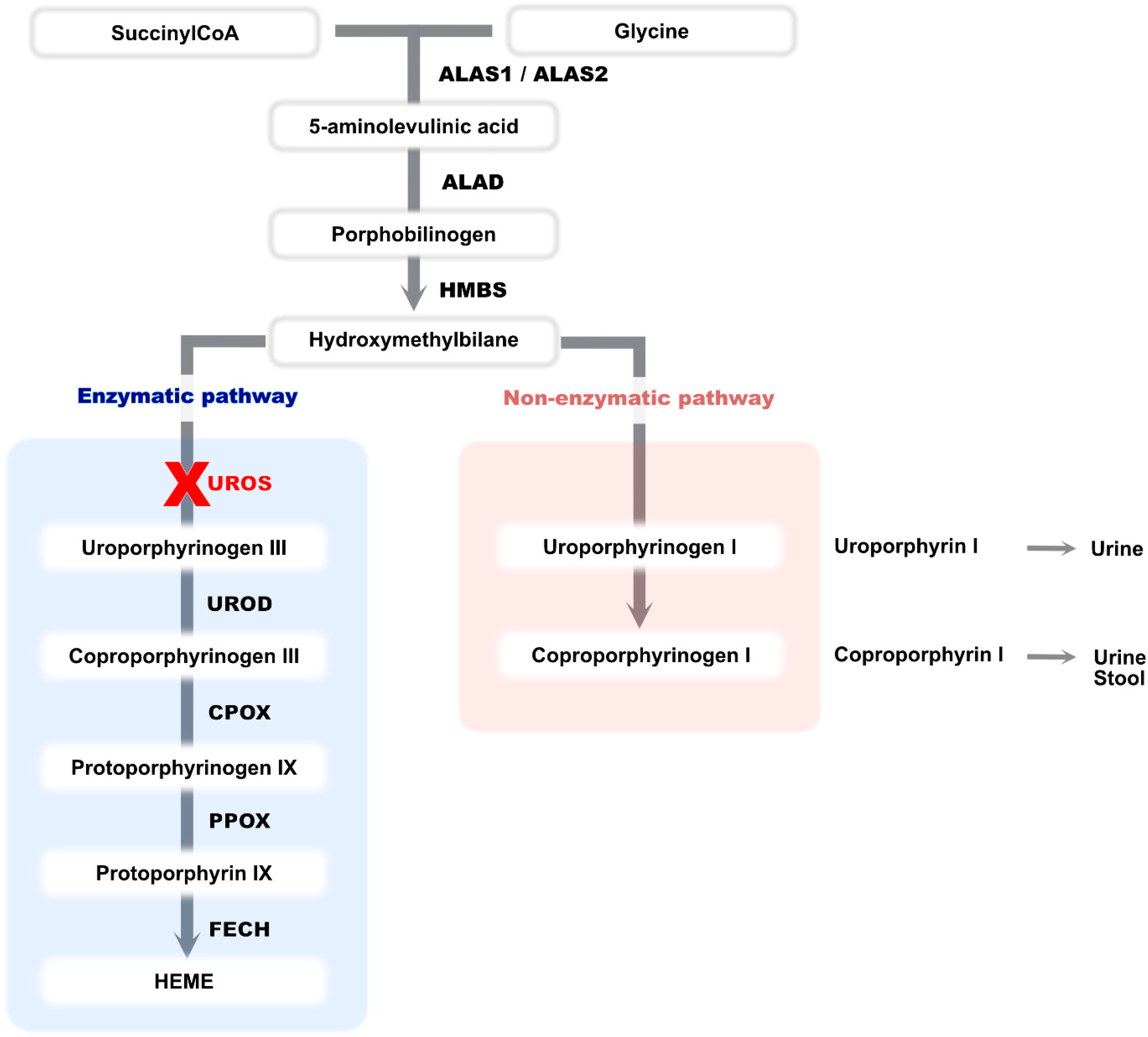
:1. Introduction
2. Patients and Methods
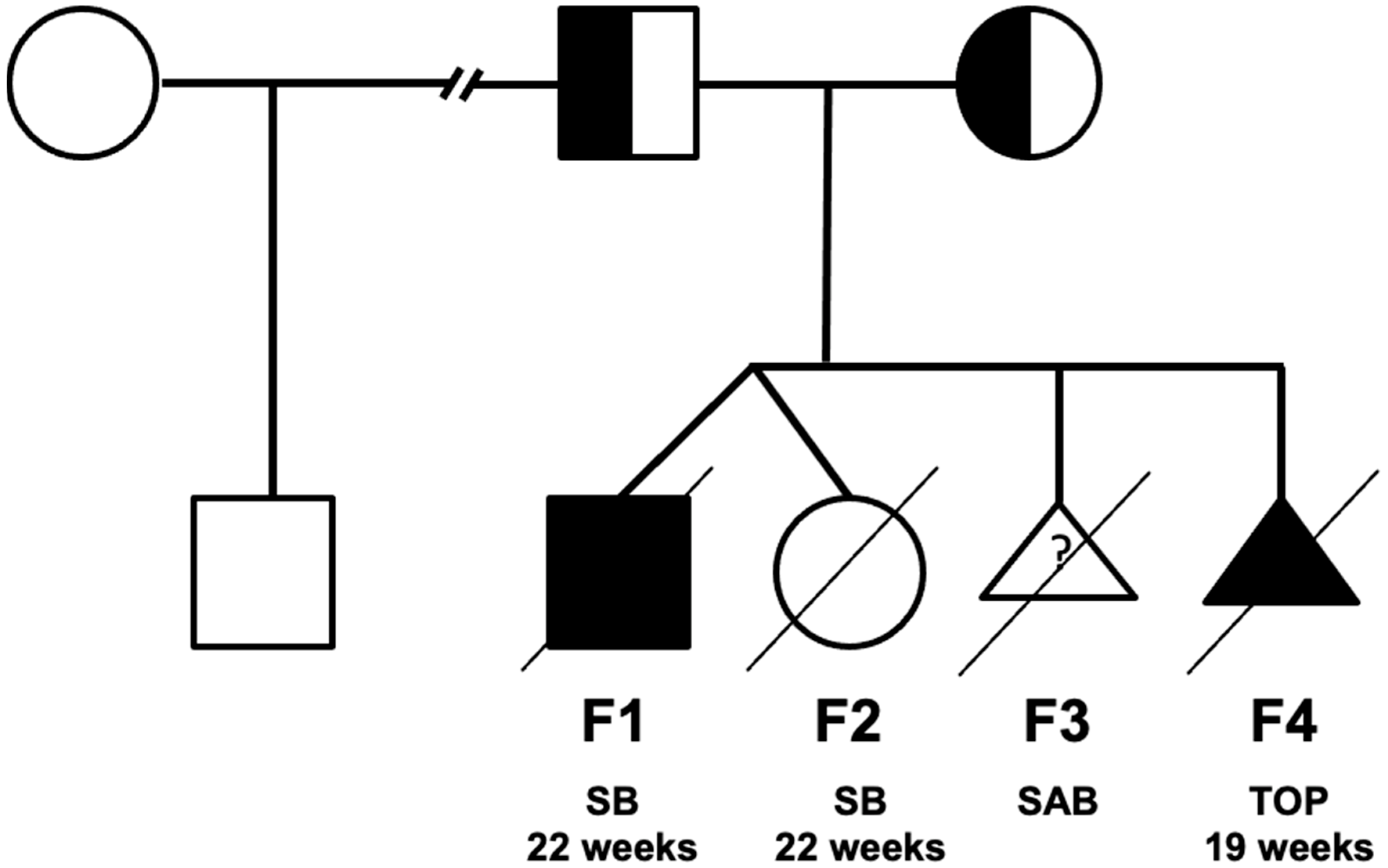
2.1. Case Description
2.2. Next Generation Sequencing
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Di Pierro, E.; Brancaleoni, V.; Granata, F. Advances in understanding the pathogenesis of congenital erythropoietic porphyria. Br. J. Haematol. 2016, 173, 365–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dsnick, R.J.; Astrin, K.H. Congenital erythropoietic porphyria: Advances in pathogenesis and treatment. Br. J. Haematol. 2002, 117, 779–795. [Google Scholar] [CrossRef] [PubMed]
- Erwin, A.L.; Desnick, R.J. Congenital erythropoietic porphyria: Recent advances. In Molecular Genetics and Metabolism; Academic Press Inc.: Cambridge, MA, USA, 2019; Volume 128, pp. 288–297. [Google Scholar]
- Solis, C.; Aizencang, G.I.; Astrin, K.H.; Bishop, D.F.; Desnick, R.J. Uroporphyrinogen iii synthase erythroid promoter mutations in adjacent gata1 and cp2 elements cause congenital erythropoietic porphyria. J. Clin. Investig. 2001, 107, 753–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandes, J.; Saudubray, J.M.; van den Berghe, G. Inborn Metabolic Diseases: Diagnosis and Treatment; Springer: Berlin/Heidelberg, Germany, 2013. [Google Scholar]
- Pannier, E.; Viot, G.; Aubry, M.C.; Grange, G.; Tantau, J.; Fallet-Bianco, C.; Muller, F.; Cabrol, D. Congenital erythropoietic porphyria (günther’s disease): Two cases with very early prenatal manifestation and cystic hygroma. Prenat. Diagn. 2003, 23, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Daïkha-Dahmane, F.; Dommergues, M.; Narcy, F.; Gubler, M.C.; Dumez, Y.; Gauthier, E.; Nordmann, Y.; Nessmann, C.; Terrasse, G.; Muller, F. Congenital erythropoietic porphyria: Prenatal diagnosis and autopsy findings in two sibling fetuses. Pediatric Dev. Pathol. 2001, 4, 180–184. [Google Scholar] [CrossRef] [PubMed]
- Lienhardt, A.; Aubard, Y.; Laroche, C.; Gilbert, B.; Bernard, P.; Massri, K.; Bouleisteix, J. A rare cause of fetal ascites: A case report of gunther’s disease. Fetal Diagn. Ther. 1999, 14, 257–261. [Google Scholar] [CrossRef] [PubMed]
- Lazebnik, N.; Lazebnik, R.S. The prenatal presentation of congenital erythropoietic porphyria: Report of two siblings with elevated maternal serum α-fetoprotein. Prenat. Diagn. 2004, 24, 282–286. [Google Scholar] [CrossRef] [PubMed]
- Verstraeten, L.; Van Regemorter, N.; Pardou, A.; de Verneuil, H.; Da Silva, V.; Rodesch, F.; Vermeylen, D.; Donner, C.; Noël, J.C.; Nordmann, Y.; et al. Biochemical diagnosis of a fatal case of günther’s disease in a newborn with hydrops foetalis. Eur. J. Clin. Chem. Clin. Biochem. 1993, 31, 121–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sudrié-Arnaud, B.; Marguet, F.; Patrier, S.; Martinovic, J.; Louillet, F.; Broux, F.; Charbonnier, F.; Dranguet, H.; Coutant, S.; Vezain, M.; et al. Metabolic causes of nonimmune hydrops fetalis: A next-generation sequencing panel as a first-line investigation. Clin. Chim. Acta 2018, 481, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Muller, E.; Brault, B.; Holmes, A.; Legros, A.; Jeannot, E.; Campitelli, M.; Rousselin, A.; Goardon, N.; Frébourg, T.; Krieger, S.; et al. Genetic profiles of cervical tumors by high-throughput sequencing for personalized medical care. Cancer Med. 2015, 4, 1484–1493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Backenroth, D.; Homsy, J.; Murillo, L.R.; Glessner, J.; Lin, E.; Brueckner, M.; Lifton, R.; Goldmuntz, E.; Chung, W.K.; Shen, Y. Canoes: Detecting rare copy number variants from whole exome sequencing data. Nucleic Acids Res. 2014, 42, e97. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the american college of medical genetics and genomics and the association for molecular pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Ged, C.; Moreau-Gaudry, F.; Richard, E.; Robert-Richard, E.; de Verneuil, H. Congenital erythropoietic porphyria: Mutation update and correlations between genotype and phenotype. Cell. Mol. Biol. 2009, 55, 53–60. [Google Scholar] [PubMed]
- Parenti, G.; Andria, G.; Ballabio, A. Lysosomal storage diseases: From pathophysiology to therapy. Annu. Rev. Med. 2015, 66, 471–486. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sudrié-Arnaud, B.; Legendre, M.; Snanoudj, S.; Pelluard, F.; Bekri, S.; Tebani, A. An Atypical Case of Congenital Erythropoietic Porphyria. Genes 2021, 12, 1828. https://doi.org/10.3390/genes12111828
Sudrié-Arnaud B, Legendre M, Snanoudj S, Pelluard F, Bekri S, Tebani A. An Atypical Case of Congenital Erythropoietic Porphyria. Genes. 2021; 12(11):1828. https://doi.org/10.3390/genes12111828
Chicago/Turabian StyleSudrié-Arnaud, Bénédicte, Marine Legendre, Sarah Snanoudj, Fanny Pelluard, Soumeya Bekri, and Abdellah Tebani. 2021. "An Atypical Case of Congenital Erythropoietic Porphyria" Genes 12, no. 11: 1828. https://doi.org/10.3390/genes12111828
APA StyleSudrié-Arnaud, B., Legendre, M., Snanoudj, S., Pelluard, F., Bekri, S., & Tebani, A. (2021). An Atypical Case of Congenital Erythropoietic Porphyria. Genes, 12(11), 1828. https://doi.org/10.3390/genes12111828