
Poking COVID-19: Insights on Genomic Constraints among Immune-Related Genes between Qatari and Italian Populations

 , , , , , , ,
, , , , , , ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Population Description
2.2. Principal Component Analysis
2.3. Genes Selection and Prioritization Analyses
2.4. WES COVID-19 Cohorts
3. Results
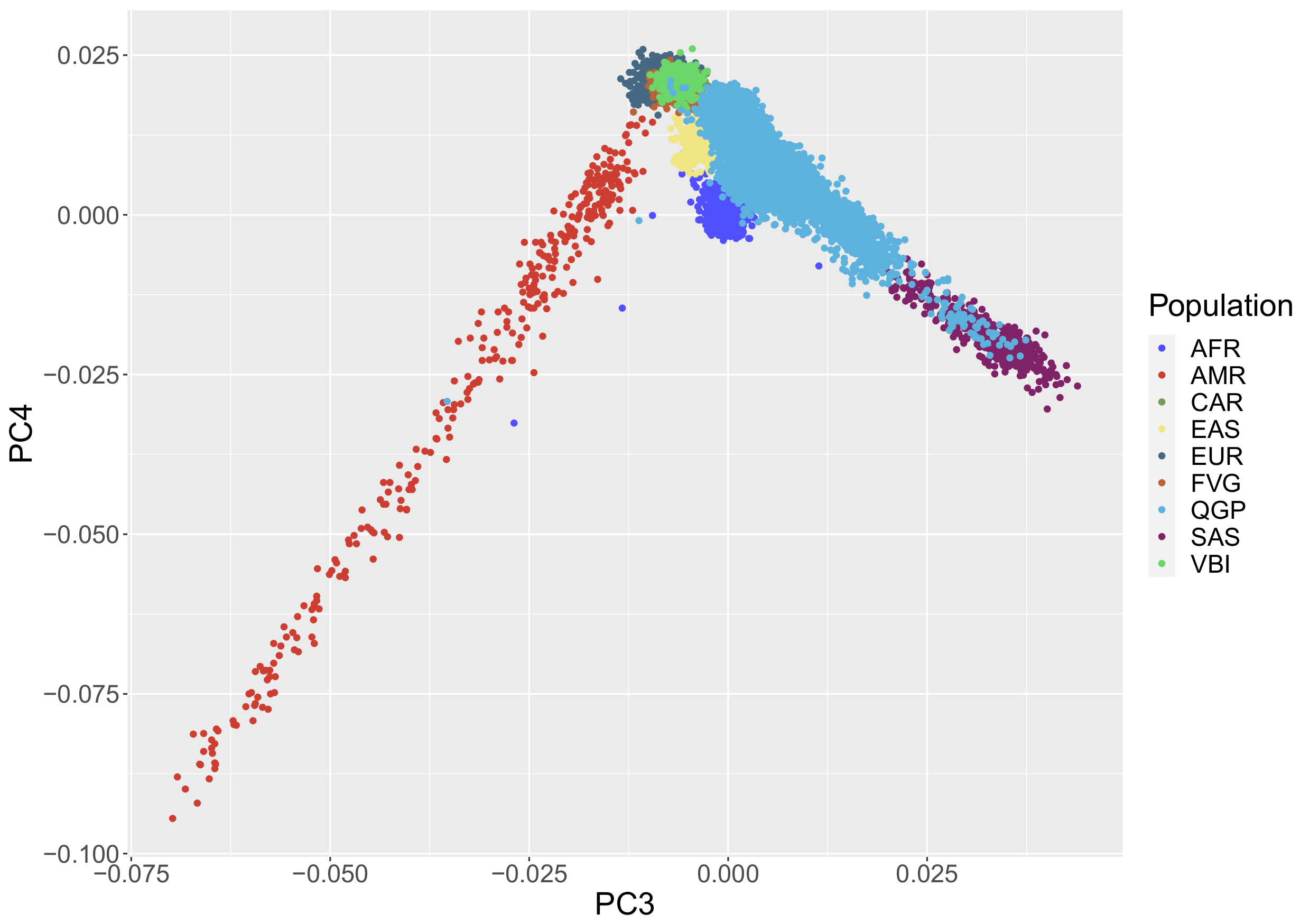
3.1. Population Stratification
3.2. Population Based Gene Prioritization
3.3. COVID-19 Cohort Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Price-Haywood, E.G.; Burton, J.; Fort, D.; Seoane, L. Hospitalization and Mortality among Black Patients and White Patients with COVID-19. N. Engl. J. Med. 2020, 382, 2534–2543. [Google Scholar] [CrossRef] [PubMed]
- Al Kuwari, H.M.; Rahim, H.F.A.; Abu-Raddad, L.J.; Abou-Samra, A.B.; Al Kanaani, Z.; Al Khal, A.; Butt, A.A.; Al Kuwari, E.; Al Marri, S.; Al Masalmani, M.; et al. Epidemiological investigation of the first 5685 cases of SARS-CoV-2 infection in Qatar, 28 February–18 April 2020. BMJ Open 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Omrani, A.S.; Almaslamani, M.A.; Daghfal, J.; Alattar, R.A.; Elgara, M.; Shaar, S.H.; Ibrahim, T.B.H.; Zaqout, A.; Bakdach, D.; Akkari, A. The first consecutive 5000 patients with Coronavirus Disease 2019 from Qatar: A nation-wide cohort study. BMC Infect. Dis. 2020, 20. [Google Scholar] [CrossRef]
- Weekly Epidemiological Update on COVID-19. Available online: https://www.who.int/publications/m/item/weekly-epidemiological-update-on-covid-19 (accessed on 31 August 2021).
- Milne, R. Societal considerations in host genome testing for COVID-19. Genet. Med. 2020, 22, 1464–1466. [Google Scholar] [CrossRef]
- Pairo-Castineira, E.; Clohisey, S.; Klaric, L.; Bretherick, A.D.; Rawlik, K.; Pasko, D.; Baillie, J.K.; Walker, S.; Parkinson, N.; Fourman, M.H.; et al. Genetic mechanisms of critical illness in COVID-19. Nature 2021, 591, 92–98. [Google Scholar] [CrossRef]
- COVID-19 Host Genetics Initiative. Mapping the human genetic architecture of COVID-19. Nature 2021. [Google Scholar] [CrossRef]
- Asano, T.; Boisson, B.; Onodi, F.; Matuozzo, D.; Moncada-Velez, M.; Zhang, P.; Meertens, L.; Bolze, A.; Casanova, J.L.; Gervais, A.; et al. X-linked recessive TLR7 deficiency in ∼1 old with life-threatening COVID-19. Sci. Immunol. 2021, 6, eabl4348. [Google Scholar] [CrossRef]
- Kousathanas, A.; Pairo-Castineira, E.; Rawlik, K.; Stuckey, A.; Odhams, C.A.; Walker, S.; Russell, C.D.; Malinauskas, T.; Millar, J.; Elliott, K.S.; et al. Whole genome sequencing identifies multiple loci for critical illness caused by COVID-19. medRxiv 2021, 15. [Google Scholar] [CrossRef]
- Degenhardt, F.; Ellinghaus, D.; Juzenas, S.; Lerga-Jaso, J.; Wendorff, M.; Maya-Miles, D.; Uellendahl-Werth, F.; ElAbd, H.; Arora, J.; Özer, O.; et al. New susceptibility loci for severe COVID-19 by detailed GWAS analysis in European populations. medRxiv 2021, 9. [Google Scholar] [CrossRef]
- The COVID-19 Host Genetics Initiative. A global initiative to elucidate the role of host genetic factors in susceptibility and severity of the SARS-CoV-2 virus pandemic. Eur. J. Hum. Genet. EJHG 2020, 28, 715–718. [Google Scholar] [CrossRef]
- Bioscience Genomics Involved in a Genetic Study on COVID-19-Bioscience Institute. Available online: https://bioinst.com/en/bioscience-genomics-involved-in-a-genetic-study-on-covid-19/ (accessed on 15 November 2021).
- Colona, V.L.; Vasilou, V.; Watt, J.; Novelli, G.; Reichardt, J.K. Update on human genetic susceptibility to COVID-19: Susceptibility to virus and response. Hum. Genom. 2021, 15. [Google Scholar] [CrossRef]
- Abu-Raddad, L.J.; Chemaitelly, H.; Ayoub, H.H.; Al Kanaani, Z.; Al Khal, A.; Al Kuwari, E.; Butt, A.A.; Coyle, P.; Jeremijenko, A.; Kaleeckal, A.H.; et al. Characterizing the Qatar advanced-phase SARS-CoV-2 epidemic. Sci. Rep. 2021, 11, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Jeremijenko, A.; Chemaitelly, H.; Ayoub, H.H.; Alishaq, M.; Abou-Samra, A.B.; Ajmi, J.A.A.A.; Ansari, N.A.A.A.; Kanaani, Z.A.; Khal, A.A.; Kuwari, E.A.; et al. Herd Immunity against Severe Acute Respiratory Syndrome Coronavirus 2 Infection in 10 Communities, Qatar-Volume 27, Number 5–May 2021-Emerging Infectious Diseases journal-CDC. Emerg. Infect. Dis. 2021, 27, 1343–1352. [Google Scholar] [CrossRef] [PubMed]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Gruber, W.C.; Marc, G.P.; Moreira, E.D.; Zerbini, C.; et al. Safety and Efficacy of the BNT162b2 mRNA COVID-19 Vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. [Google Scholar] [CrossRef] [PubMed]
- Baden, L.R.; El Sahly, H.M.; Essink, B.; Kotloff, K.; Frey, S.; Novak, R.; Zaks, T.; Diemert, D.; Spector, S.A.; Rouphael, N.; et al. Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. N. Engl. J. Med. 2021, 384, 403–416. [Google Scholar] [CrossRef]
- Voysey, M.; Clemens, S.A.C.; Madhi, S.A.; Weckx, L.Y.; Folegatti, P.M.; Aley, P.K.; Angus, B.; Baillie, V.L.; Barnabas, S.L.; Bhorat, Q.E.; et al. Safety and efficacy of the ChAdOx1 nCoV-19 vaccine (AZD1222) against SARS-CoV-2: An interim analysis of four randomised controlled trials in Brazil, South Africa, and the UK. Lancet 2021, 397, 99–111. [Google Scholar] [CrossRef]
- Logunov, D.Y.; Dolzhikova, I.V.; Shcheblyakov, D.V.; Tukhvatulin, A.I.; Zubkova, O.V.; Dzharullaeva, A.S.; Kovyrshina, A.V.; Lubenets, N.L.; Grousova, D.M.; Erokhova, A.S.; et al. Safety and efficacy of an rAd26 and rAd5 vector-based heterologous prime-boost COVID-19 vaccine: An interim analysis of a randomised controlled phase 3 trial in Russia. Lancet 2021, 397, 671–681. [Google Scholar] [CrossRef]
- Stolfi, P.; Manni, L.; Soligo, M.; Vergni, D.; Tieri, P. Designing a Network Proximity-Based Drug Repurposing Strategy for COVID-19. Front. Cell Dev. Biol. 2020, 0, 1021. [Google Scholar] [CrossRef]
- Mbarek, H.; Gandhi, G.D.; Selvaraj, S.; Al-Muftah, W.; Badji, R.; Al-Sarraj, Y.; Saad, C.; Darwish, D.; Alvi, M.; Fadl, T.; et al. Qatar Genome: Insights on Genomics from the Middle East. medRxiv 2021. [Google Scholar] [CrossRef]
- Al Thani, A.; Fthenou, E.; Paparrodopoulos, S.; Al Marri, A.; Shi, Z.; Qafoud, F.; Afifi, N. Qatar Biobank Cohort Study: Study Design and First Results. Am. J. Epidemiol. 2019, 188, 1420–1433. [Google Scholar] [CrossRef]
- Razali, R.M.; Rodriguez-Flores, J.; Ghorbani, M.; Naeem, H.; Aamer, W.; Aliyev, E.; Jubran, A.; Clark, A.G.; Fakhro, K.A.; Mokrab, Y. Thousands of Qatari genomes inform human migration history and improve imputation of Arab haplotypes. Nat. Commun. 2021, 12, 1–16. [Google Scholar] [CrossRef]
- Cocca, M.; Barbieri, C.; Concas, M.P.; Robino, A.; Brumat, M.; Gandin, I.; Trudu, M.; Sala, C.F.; Vuckovic, D.; Girotto, G.; et al. A bird’s-eye view of Italian genomic variation through whole-genome sequencing. Eur. J. Hum. Genet. 2020, 28, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Latini, A.; Agolini, E.; Novelli, A.; Borgiani, P.; Giannini, R.; Gravina, P.; Smarrazzo, A.; Dauri, M.; Andreoni, M.; Rogliani, P.; et al. COVID-19 and genetic variants of protein involved in the SARS-CoV-2 entry into the host cells. Genes 2020, 11, 1010. [Google Scholar] [CrossRef] [PubMed]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manichaikul, A.; Mychaleckyj, J.C.; Rich, S.S.; Daly, K.; Sale, M.; Chen, W.M. Robust relationship inference in genome-wide association studies. Bioinformatics 2010, 26, 2867–2873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. GigaScience 2015, 4, 7. [Google Scholar] [CrossRef] [PubMed]
- Auton, A.; Abecasis, G.R.; Altshuler, D.M.; Durbin, R.M.; Bentley, D.R.; Chakravarti, A.; Clark, A.G.; Donnelly, P.; Eichler, E.E.; Flicek, P.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef] [Green Version]
- QIAGEN-Inc. Ingenuity Variant Analysis (IVA). Available online: https://www.qiagenbioinformatics.com/products/ingenuity-variant-analysis (accessed on 15 November 2021).
- Martin, A.R.; Williams, E.; Foulger, R.E.; Leigh, S.; Daugherty, L.C.; Niblock, O.; Leong, I.U.S.; Smith, K.R.; Gerasimenko, O.; Haraldsdottir, E.; et al. PanelApp crowdsources expert knowledge to establish consensus diagnostic gene panels. Nat. Genet. 2019, 51, 1560–1565. [Google Scholar] [CrossRef]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [Green Version]
- Petrovski, S.; Wang, Q.; Heinzen, E.L.; Allen, A.S.; Goldstein, D.B. Genic Intolerance to Functional Variation and the Interpretation of Personal Genomes. PLoS Genet. 2013, 9, e1003709. [Google Scholar] [CrossRef]
- Mezzavilla, M.; Cocca, M.; Guidolin, F.; Gasparini, P. A population-based approach for gene prioritization in understanding complex traits. Hum. Genet. 2020, 139, 647–655. [Google Scholar] [CrossRef] [PubMed]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Esko, T.; Mezzavilla, M.; Nelis, M.; Borel, C.; Debniak, T.; Jakkula, E.; Julia, A.; Karachanak, S.; Khrunin, A.; Kisfali, P.; et al. Genetic characterization of northeastern Italian population isolates in the context of broader European genetic diversity. Eur. J. Hum. Genet. 2013, 21, 659–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thareja, G.; Al-Sarraj, Y.; Belkadi, A.; Almotawa, M.; Ismail, S.; Al-Muftah, W.; Badji, R.; Mbarek, H.; Darwish, D.; Fadl, T.; et al. Whole genome sequencing in the Middle Eastern Qatari population identifies genetic associations with 45 clinically relevant traits. Nat. Commun. 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Ehre, C.; Worthington, E.N.; Liesman, R.M.; Grubb, B.R.; Barbier, D.; O’Neal, W.K.; Sallenave, J.M.; Pickles, R.J.; Boucher, R.C. Overexpressing mouse model demonstrates the protective role of Muc5ac in the lungs. Proc. Natl. Acad. Sci. USA 2012, 109, 16528–16533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, W.; Liu, X.; Wang, T.; Liu, F.; Zhu, A.; Lin, Y.; Luo, J.; Ye, F.; He, J.; Zhao, J.; et al. Elevated MUC1 and MUC5AC mucin protein levels in airway mucus of critical ill COVID-19 Patients. J. Med. Virol. 2021, 93, 582–584. [Google Scholar] [CrossRef]
- Trincone, A.; Schwegmann-Weßels, C. Looking for a needle in a haystack: Cellular proteins that may interact with the tyrosine-based sorting signal of the TGEV S protein. Virus Res. 2015, 202, 3–11. [Google Scholar] [CrossRef]
- Chakrabarty, B.; Das, D.; Bulusu, G.; Roy, A. Network-Based Analysis of Fatal Comorbidities of COVID-19 and Potential Therapeutics. IEEE/ACM Trans. Comput. Biol. Bioinform. 2021, 18, 1271–1280. [Google Scholar] [CrossRef] [PubMed]
- Valentin, V.; Bervar, J.F.; Vincent-Delorme, C.; Smol, T.; Wemeau, L.; Remy, M.; Le Rouzic, O.; Chenivesse, C. Filamin A Mutations: A New Cause of Unexplained Emphysema in Adults? Chest 2021, 159, e131–e135. [Google Scholar] [CrossRef]
- Jehle, A.W.; Gardai, S.J.; Li, S.; Linsel-Nitschke, P.; Morimoto, K.; Janssen, W.J.; Vandivier, R.W.; Wang, N.; Greenberg, S.; Dale, B.M.; et al. ATP-binding cassette transporter A7 enhances phagocytosis of apoptotic cells and associated ERK signaling in macrophages. J. Cell Biol. 2006, 174, 547–556. [Google Scholar] [CrossRef] [Green Version]
- Iwamoto, N.; Abe-Dohmae, S.; Sato, R.; Yokoyama, S. ABCA7 expression is regulated by cellular cholesterol through the SREBP2 pathway and associated with phagocytosis. J. Lipid Res. 2006, 47, 1915–1927. [Google Scholar] [CrossRef] [Green Version]
- Oughtred, R.; Rust, J.; Chang, C.; Breitkreutz, B.J.; Stark, C.; Willems, A.; Boucher, L.; Leung, G.; Kolas, N.; Zhang, F.; et al. The BioGRID database: A comprehensive biomedical resource of curated protein, genetic, and chemical interactions. Protein Sci. 2021, 30, 187–200. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Hou, Y.; Shen, J.; Mehra, R.; Kallianpur, A.; Culver, D.A.; Gack, M.U.; Farha, S.; Zein, J.; Comhair, S.; et al. A network medicine approach to investigation and population-based validation of disease manifestations and drug repurposing for COVID-19. PLoS Biol. 2020, 18. [Google Scholar] [CrossRef] [PubMed]
- Woodruff, T.M.; Shukla, A.K. The Complement C5a-C5aR1 GPCR Axis in COVID-19 Therapeutics. Trends Immunol. 2020. [Google Scholar] [CrossRef]
- Lee, M.E.; Chang, Y.; Ahmadinejad, N.; Johnson-Agbakwu, C.E.; Bailey, C.; Liu, L. COVID-19 Mortality is Associated with Impaired Innate Immunity in Pre-existing Health Conditions. bioRxiv 2021. [Google Scholar] [CrossRef]
- Cao, Y.; Li, L.; Feng, Z.; Wan, S.; Huang, P.; Sun, X.; Wen, F.; Huang, X.; Ning, G.; Wang, W. Comparative genetic analysis of the novel coronavirus (2019-nCoV/SARS-CoV-2) receptor ACE2 in different populations. Cell Discov. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Secolin, R.; de Araujo, T.K.; Gonsales, M.C.; Rocha, C.S.; Naslavsky, M.; Marco, L.D.; Bicalho, M.A.; Vazquez, V.L.; Zatz, M.; Silva, W.A.; et al. Genetic variability in COVID-19-related genes in the Brazilian population. Hum. Genome Var. 2021, 8. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| DSC Score | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Transcript ID | Gene Name | QGP | CAR | FVG | VBI | EUR | AFR | SAS | Comparison |
| ENST00000369850 | FLNA | 3.854 | −2.435 | 0.272 | −2.510 | −2.399 | −2.166 | −1.879 | C5 |
| ENST00000350763 | TNC | 3.370 | −3.792 | 1.388 | −0.631 | 2.666 | 1.838 | 2.187 | C4 |
| ENST00000389048 | ALK | 2.575 | 3.651 | 0.290 | −4.098 | 3.388 | 3.212 | 2.852 | C4 |
| ENST00000263094 | ABCA7 | 2.566 | −0.433 | −2.168 | 0.071 | 3.020 | 2.681 | 2.150 | C4 |
| ENST00000647814 | ABCC2 | 2.528 | −3.466 | 0.467 | 3.004 | 2.562 | 2.877 | 2.508 | C4 |
| ENST00000621226 | MUC5AC | 2.435 | −2.404 | −2.017 | −2.892 | 3.477 | 3.500 | 3.032 | C4 |
| ENST00000634891 | RYR3 | 2.229 | −3.377 | −1.554 | −3.586 | −2.431 | 2.639 | −3.449 | C8 |
| ENST00000542267 | FBXL17 | 2.026 | −1.232 | 0.180 | −2.658 | 3.086 | 0.266 | 2.477 | C4 |
| ENST00000589042 | TTN | −2.242 | −2.595 | 3.411 | 4.584 | −2.388 | −1.965 | 3.498 | C1 |
| ENST00000357387 | RICTOR | −2.369 | −2.206 | −0.033 | 2.407 | 2.181 | 1.284 | −4.070 | C7 |
| ENST00000561890 | MUC22 | −2.472 | −1.682 | −1.632 | 2.156 | −2.562 | −2.191 | −2.425 | C3 |
| ENST00000336596 | EPHA3 | −3.001 | 2.139 | −3.421 | −1.535 | −3.704 | 1.921 | −2.814 | C3 |
| ENST00000648947 | INO80 | −3.444 | −1.309 | 2.424 | −2.928 | −3.439 | −2.692 | −0.727 | C3 |
| ENST00000389484 | LRP1B | −4.888 | −4.916 | 3.192 | 2.602 | −4.245 | −2.136 | 3.231 | C1 |
| SSC Score | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Transcript ID | Gene Name | QGP | CAR | FVG | VBI | EUR | AFR | SAS | Comparison |
| ENST00000378473 | PLCB4 | −4.524 | 2.917 | −3.907 | −0.413 | −4.272 | −2.873 | −3.079 | C3 |
| ENST00000366574 | RYR2 | −4.347 | 3.792 | −4.694 | −5.053 | −3.704 | −2.318 | −2.087 | C3 |
| ENST00000315872 | ROCK2 | −3.680 | 3.822 | −2.372 | −1.149 | −4.160 | −3.248 | −3.665 | C3 |
| ENST00000361445 | MTOR | −3.371 | −0.975 | 0.100 | 2.659 | −3.378 | −4.276 | −3.888 | C3 |
| ENST00000358691 | HELZ | −3.131 | 4.249 | 1.658 | −3.729 | −3.437 | −2.929 | 3.413 | C1 |
| ENST00000355286 | EYA4 | −3.000 | −1.900 | 2.671 | −1.486 | −2.087 | −2.589 | −0.756 | C3 |
| ENST00000381501 | TEC | −2.996 | −2.615 | −1.656 | 3.085 | −2.497 | −2.427 | −0.767 | C3 |
| ENST00000265382 | PIP5K1B | −2.952 | 2.574 | −0.576 | −2.746 | −3.246 | −3.197 | −1.583 | C3 |
| ENST00000359015 | MAP3K5 | −2.758 | 2.108 | 0.850 | 1.431 | −3.555 | −2.472 | −3.293 | C3 |
| ENST00000335670 | RORA | −2.586 | −2.953 | 1.176 | 2.228 | −2.499 | −2.743 | −0.526 | C3 |
| ENST00000370056 | VAV3 | −2.523 | 3.467 | 1.312 | 1.038 | −3.282 | −2.646 | −1.406 | C3 |
| ENST00000381298 | IL6ST | −2.522 | −1.224 | 3.859 | 2.542 | −2.120 | −1.466 | −2.644 | C3 |
| ENST00000432237 | CD163 | −2.506 | −1.404 | 2.793 | −2.306 | −2.419 | −0.629 | −2.156 | C3 |
| ENST00000392552 | GPR155 | −2.338 | −1.261 | −1.336 | 2.338 | −2.417 | −1.608 | −2.585 | C3 |
| ENST00000382292 | SACS | −2.324 | −4.408 | 3.917 | 2.284 | −3.530 | −2.726 | −2.082 | C3 |
| ENST00000392132 | XRCC5 | −2.176 | −2.147 | 2.722 | −1.257 | −2.673 | −2.107 | −1.787 | C3 |
| ENST00000313708 | EBF1 | −2.068 | 2.253 | −1.422 | −0.914 | −2.980 | −1.665 | −3.222 | C3 |
| ENST00000400841 | CRLF2 | 2.036 | −1.347 | −1.496 | −2.083 | 2.581 | 2.185 | 1.052 | C4 |
| ENST00000369850 | FLNA | 2.058 | −3.158 | −0.860 | −4.025 | −3.073 | −3.351 | −3.097 | C5 |
| ENST00000344327 | TRPC6 | 2.062 | −3.776 | −2.671 | −2.711 | −3.382 | 0.278 | −2.242 | C5 |
| ENST00000263317 | NOX4 | 2.225 | −2.716 | −2.554 | −2.717 | 2.134 | 3.532 | 3.770 | C4 |
| ENST00000403662 | CSF2RB | 2.237 | −2.363 | 1.702 | 0.620 | 2.613 | 0.319 | 2.782 | C4 |
| ENST00000297494 | NOS3 | 2.243 | 1.436 | −2.178 | −0.851 | 2.109 | 2.460 | 2.455 | C4 |
| ENST00000295598 | ATP1A1 | 2.258 | −2.679 | 0.547 | 0.930 | −2.204 | −1.886 | −2.448 | C5 |
| ENST00000085219 | CD22 | 2.288 | 0.576 | 0.028 | −2.311 | 2.368 | −0.600 | 2.142 | C4 |
| ENST00000305877 | BCR | 2.397 | −1.338 | 2.028 | −3.021 | 3.994 | 2.923 | 3.631 | C4 |
| ENST00000333149 | TRIM50 | 2.501 | 2.275 | 1.138 | −2.022 | 2.271 | 1.372 | 3.197 | C4 |
| ENST00000271332 | CELSR2 | 2.522 | 3.651 | −2.581 | 2.455 | 2.443 | −2.129 | 2.936 | C2 |
| ENST00000447648 | TECPR1 | 2.666 | −2.351 | 1.822 | −1.669 | 2.777 | 3.213 | −0.027 | C4 |
| ENST00000324856 | ARID1A | 3.434 | −3.120 | −2.683 | −1.845 | −2.085 | −2.053 | 0.835 | C5 |
| ENST00000263094 | ABCA7 | 3.796 | −1.601 | −2.581 | 1.004 | 2.591 | 3.325 | 3.998 | C4 |
| ENST00000372923 | DNM1 | 3.941 | −2.514 | −1.222 | −0.710 | −2.066 | −1.575 | −2.077 | C5 |
| ENST00000621226 | MUC5AC | 3.965 | −3.705 | −3.751 | −4.601 | 3.679 | 3.267 | 4.244 | C4 |
| ENST00000533211 | SPTBN2 | 4.531 | −2.266 | 1.209 | −1.893 | 2.589 | 2.191 | 2.778 | C4 |
| ENST00000529681 | MUC5B | 4.744 | 3.085 | 1.483 | −2.070 | 4.884 | 4.396 | 5.095 | C4 |
| DSC Score | SSC Score | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Transcript ID | Gene Name | QGP | CAR | FVG | VBI | EUR | AFR | SAS | QGP | CAR | FVG | VBI | EUR | AFR | SAS |
| ENST00000369850 | FLNA | 3.854 | −2.435 | 0.272 | −2.510 | −2.399 | −2.166 | −1.879 | 2.058 | −3.158 | −0.860 | −4.025 | −3.073 | −3.351 | −3.097 |
| ENST00000263094 | ABCA7 | 2.566 | −0.433 | −2.168 | 0.071 | 3.020 | 2.681 | 2.150 | 3.796 | −1.601 | −2.581 | 1.004 | 2.591 | 3.325 | 3.998 |
| ENST00000621226 | MUC5AC | 2.435 | −2.404 | −2.017 | −2.892 | 3.477 | 3.500 | 3.032 | 3.965 | −3.705 | −3.751 | −4.601 | 3.679 | 3.267 | 4.244 |
| Transcript ID | Gene Name | Cohort | p-Value Whole Gene | p-Value CDS Regions |
|---|---|---|---|---|
| ENST00000369850 | FLNA | CAR | 0.630140 | 0.409653 |
| FVG | 0.046901 | 0.316565 | ||
| VBI | 0.000458 | 0.013015 | ||
| QGP | 0.000028 | 0.039803 | ||
| EUR | 0.312323 | 0.786342 | ||
| AFR | 0.878006 | 0.787767 | ||
| SAS | 0.200408 | 0.813561 | ||
| ENST00000263094 | ABCA7 | CAR | 3.2746 × 10−11 | 2.5959 × 10−7 |
| FVG | 3.1278 × 10−23 | 1.0535 × 10−17 | ||
| VBI | 7.1607 × 10−21 | 2.0060 × 10−16 | ||
| QGP | 6.2413 × 10−63 | 1.5606 × 10−40 | ||
| EUR | 4.4467 × 10−10 | 1.0966 × 10−8 | ||
| AFR | 1.7360 × 10−8 | 5.3713 × 10−9 | ||
| SAS | 2.3435 × 10−4 | 3.7924 × 10−6 | ||
| ENST00000621226 | MUC5AC | CAR | 7.4274 × 10−12 | 8.4692 × 10−11 |
| FVG | 1.8836 × 10−31 | 2.7623 × 10−24 | ||
| VBI | 2.3148 × 10−36 | 3.2118 × 10−30 | ||
| QGP | 1.5512 × 10−76 | 1.2101 × 10−64 | ||
| EUR | 3.3142 × 10−6 | 4.0071 × 10−8 | ||
| AFR | 2.9701 × 10−8 | 1.0241 × 10−10 | ||
| SAS | 7.2316 × 10−3 | 9.9471 × 10−5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mbarek, H.; Cocca, M.; Al-Sarraj, Y.; Saad, C.; Mezzavilla, M.; AlMuftah, W.; Cocciadiferro, D.; Novelli, A.; Quinti, I.; AlTawashi, A.; et al. Poking COVID-19: Insights on Genomic Constraints among Immune-Related Genes between Qatari and Italian Populations. Genes 2021, 12, 1842. https://doi.org/10.3390/genes12111842
Mbarek H, Cocca M, Al-Sarraj Y, Saad C, Mezzavilla M, AlMuftah W, Cocciadiferro D, Novelli A, Quinti I, AlTawashi A, et al. Poking COVID-19: Insights on Genomic Constraints among Immune-Related Genes between Qatari and Italian Populations. Genes. 2021; 12(11):1842. https://doi.org/10.3390/genes12111842
Chicago/Turabian StyleMbarek, Hamdi, Massimiliano Cocca, Yasser Al-Sarraj, Chadi Saad, Massimo Mezzavilla, Wadha AlMuftah, Dario Cocciadiferro, Antonio Novelli, Isabella Quinti, Azza AlTawashi, and et al. 2021. "Poking COVID-19: Insights on Genomic Constraints among Immune-Related Genes between Qatari and Italian Populations" Genes 12, no. 11: 1842. https://doi.org/10.3390/genes12111842
APA StyleMbarek, H., Cocca, M., Al-Sarraj, Y., Saad, C., Mezzavilla, M., AlMuftah, W., Cocciadiferro, D., Novelli, A., Quinti, I., AlTawashi, A., Salvaggio, S., AlThani, A., Novelli, G., & Ismail, S. I. (2021). Poking COVID-19: Insights on Genomic Constraints among Immune-Related Genes between Qatari and Italian Populations. Genes, 12(11), 1842. https://doi.org/10.3390/genes12111842