
Short Time-Series Expression Transcriptome Data Reveal the Gene Expression Patterns of Dairy Cow Mammary Gland as Milk Yield Decreased Process

 ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Milk Composition Detection and Histological Examination
2.3. Transcriptome Sequencing
2.4. Gene Expression Level Analysis
2.5. Bioinformatics Analysis of Differentially Expressed Genes
2.6. Short Time-Series Expression Miner Analysis
2.7. Validation of Sequencing Data by qRT-PCR
2.8. Statistical Analysis
3. Results
3.1. Daily Milk Yield, Milk Composition, and Somatic Cell Count
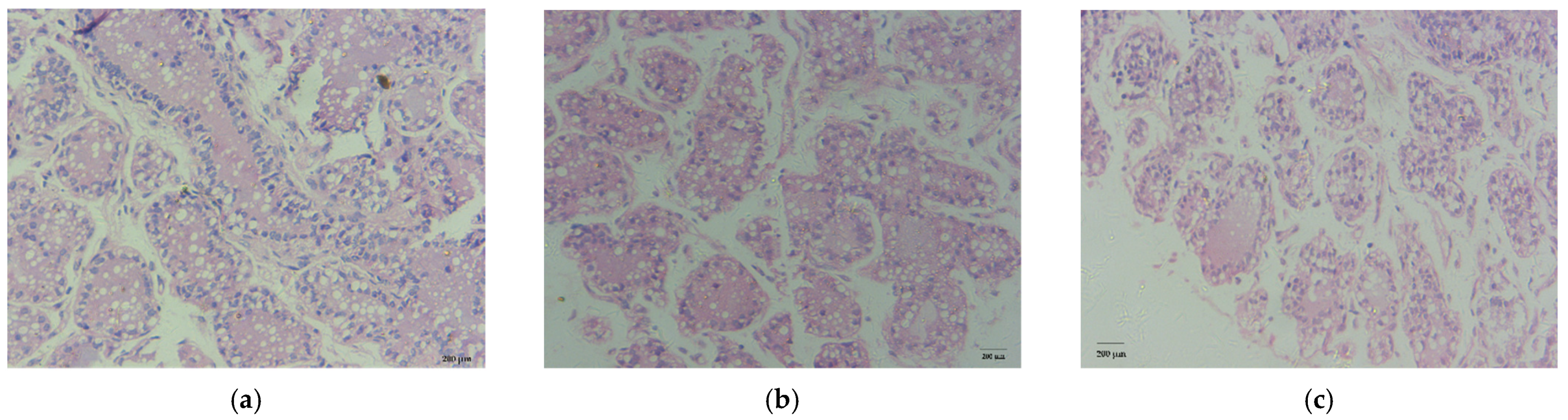
3.2. Histological Observation
3.3. Transcriptome Sequencing Results and Quality Control
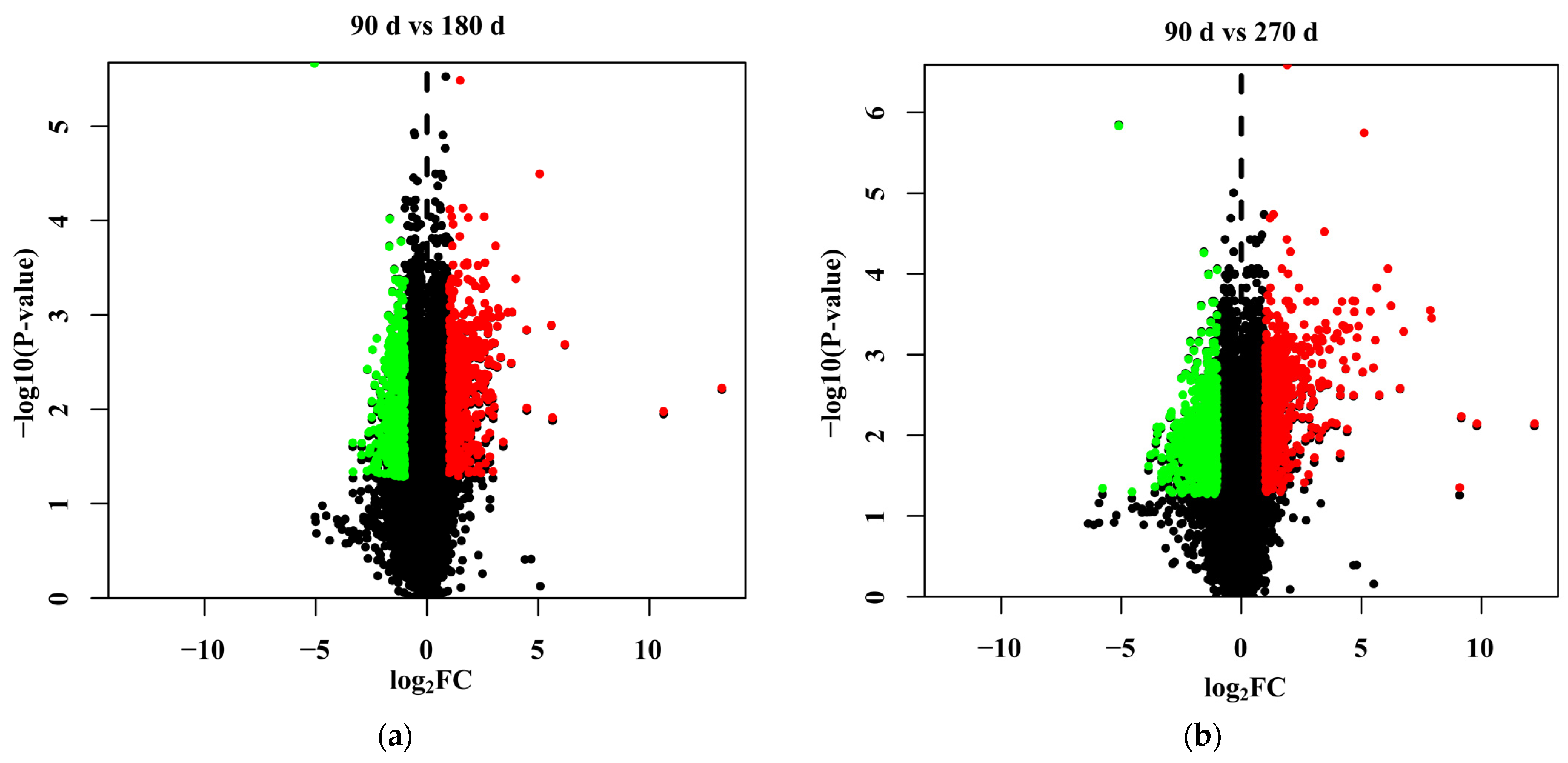
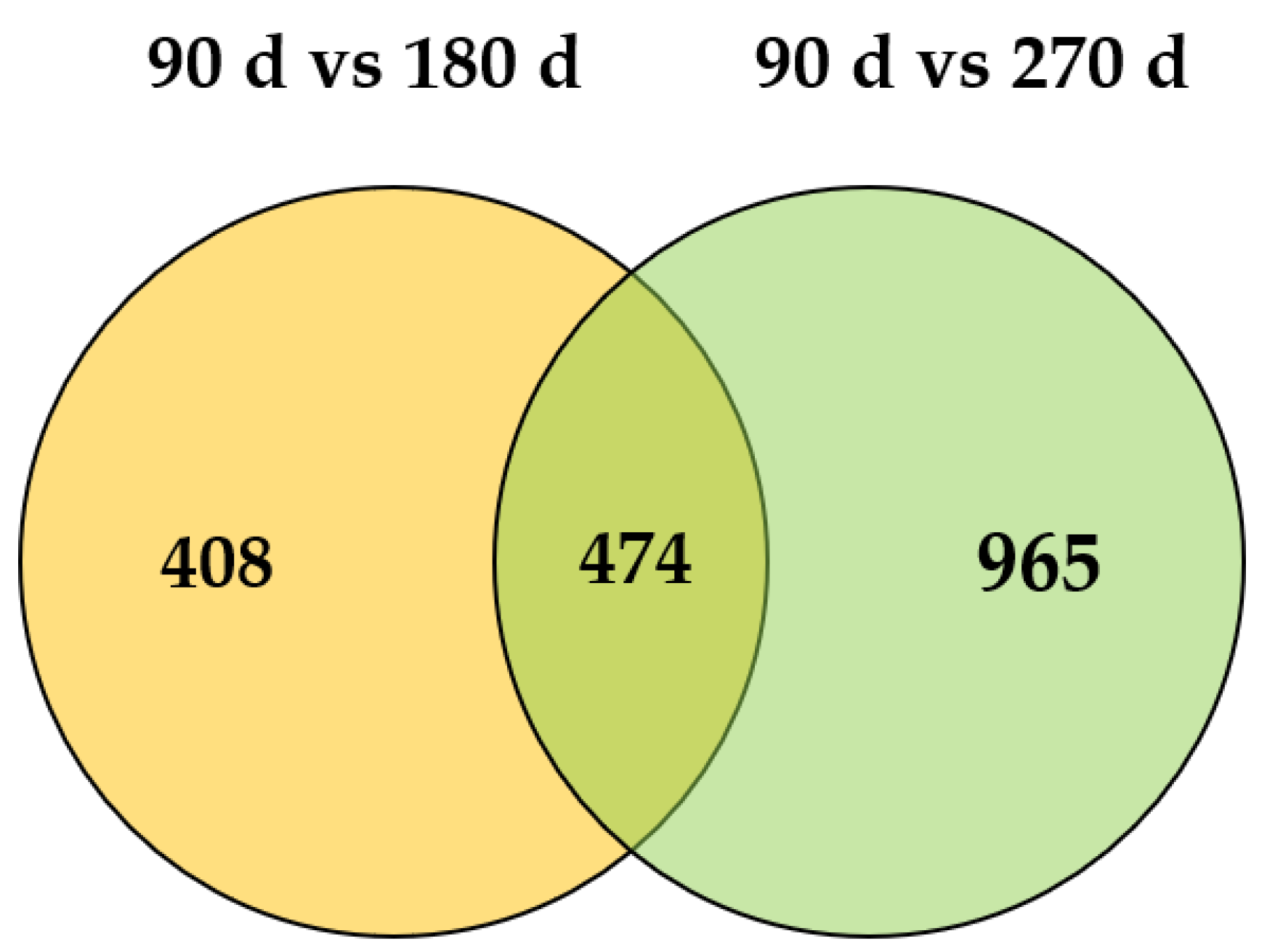
3.4. Identification of Differentially Expressed Genes
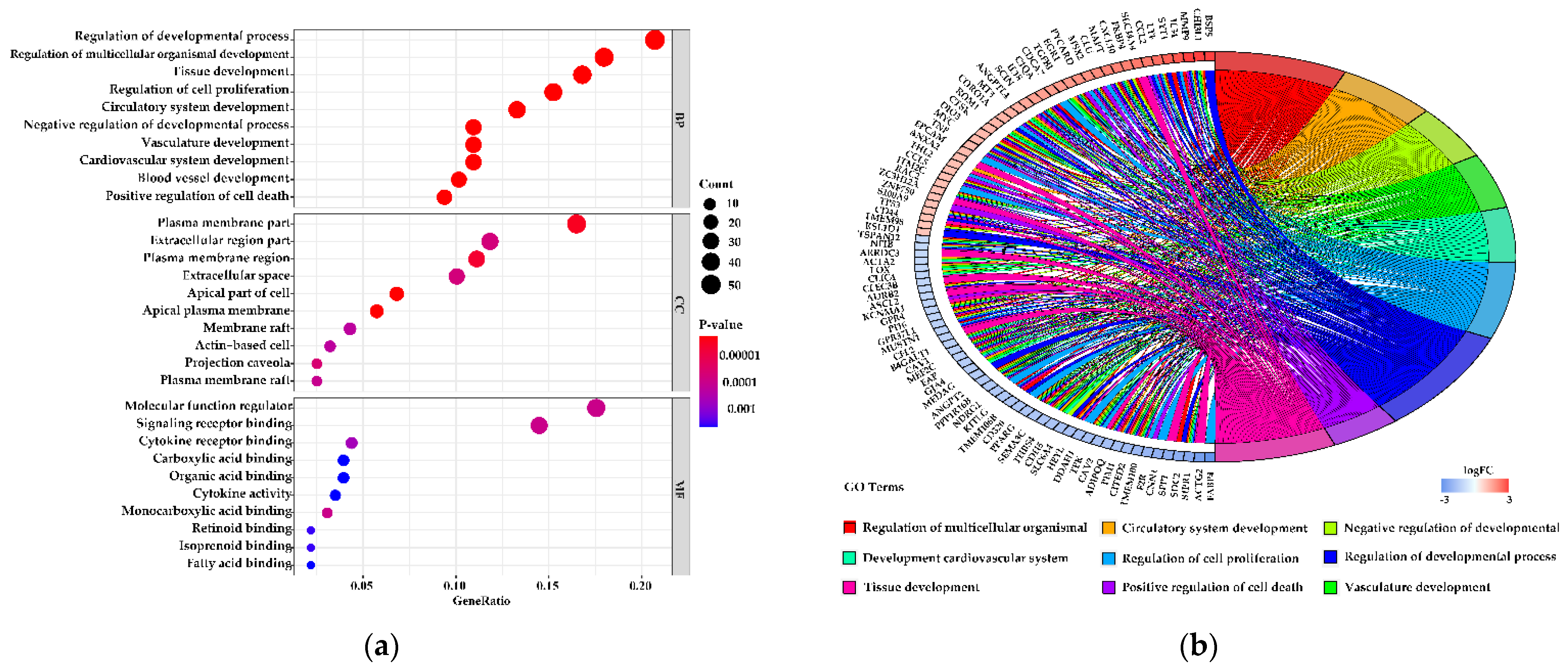
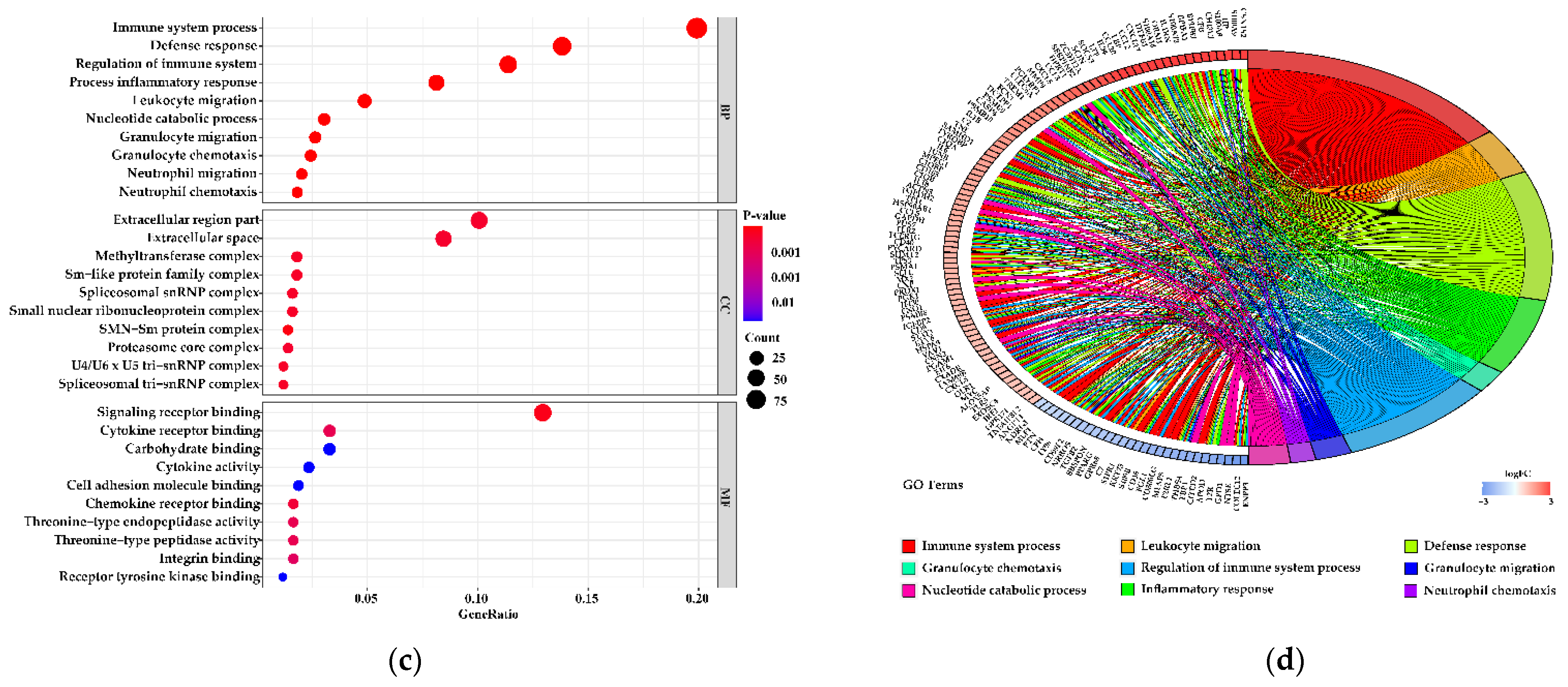
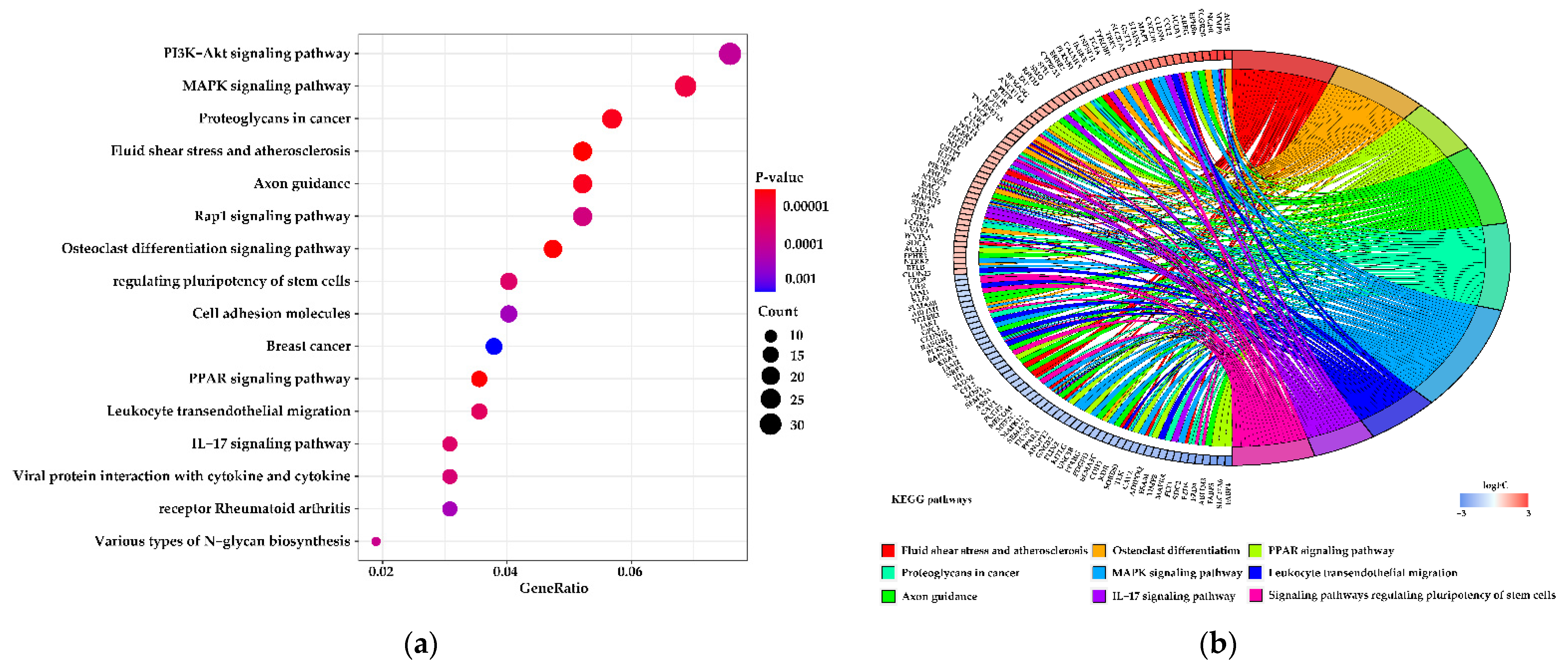
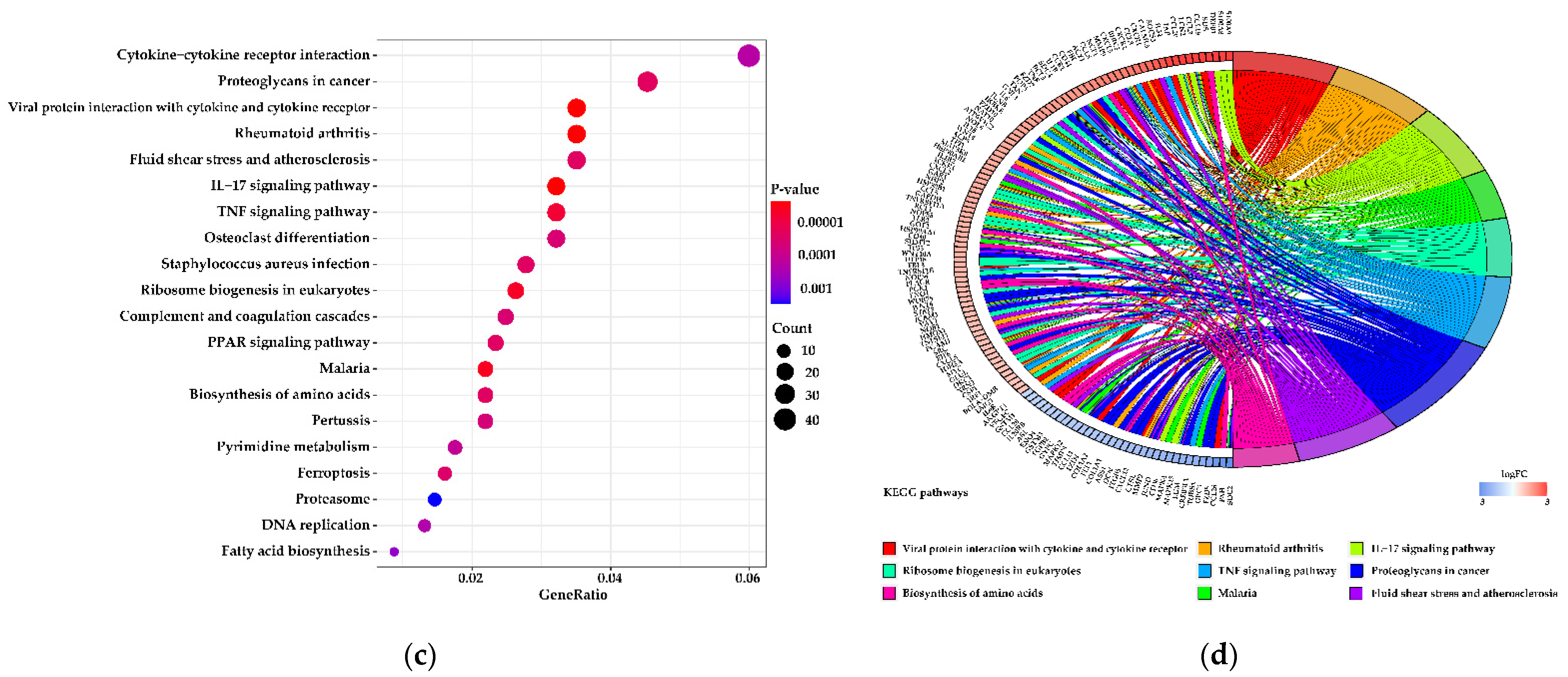
3.5. Functions of the Differentially Expressed Genes
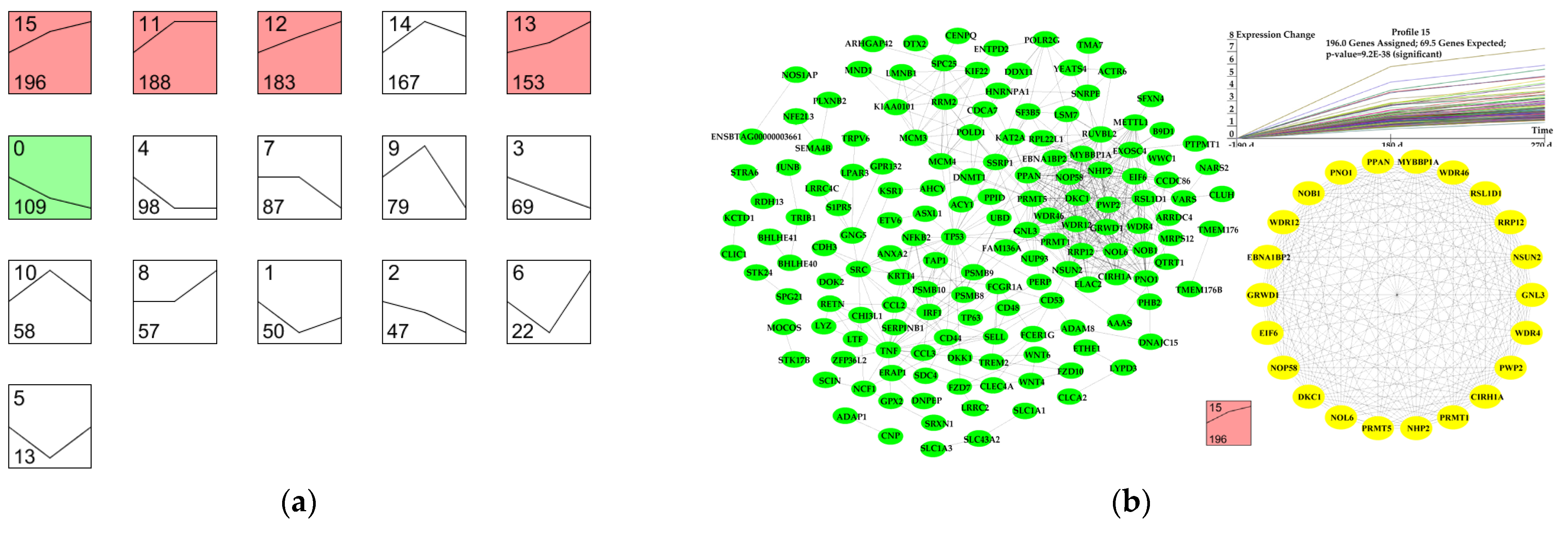
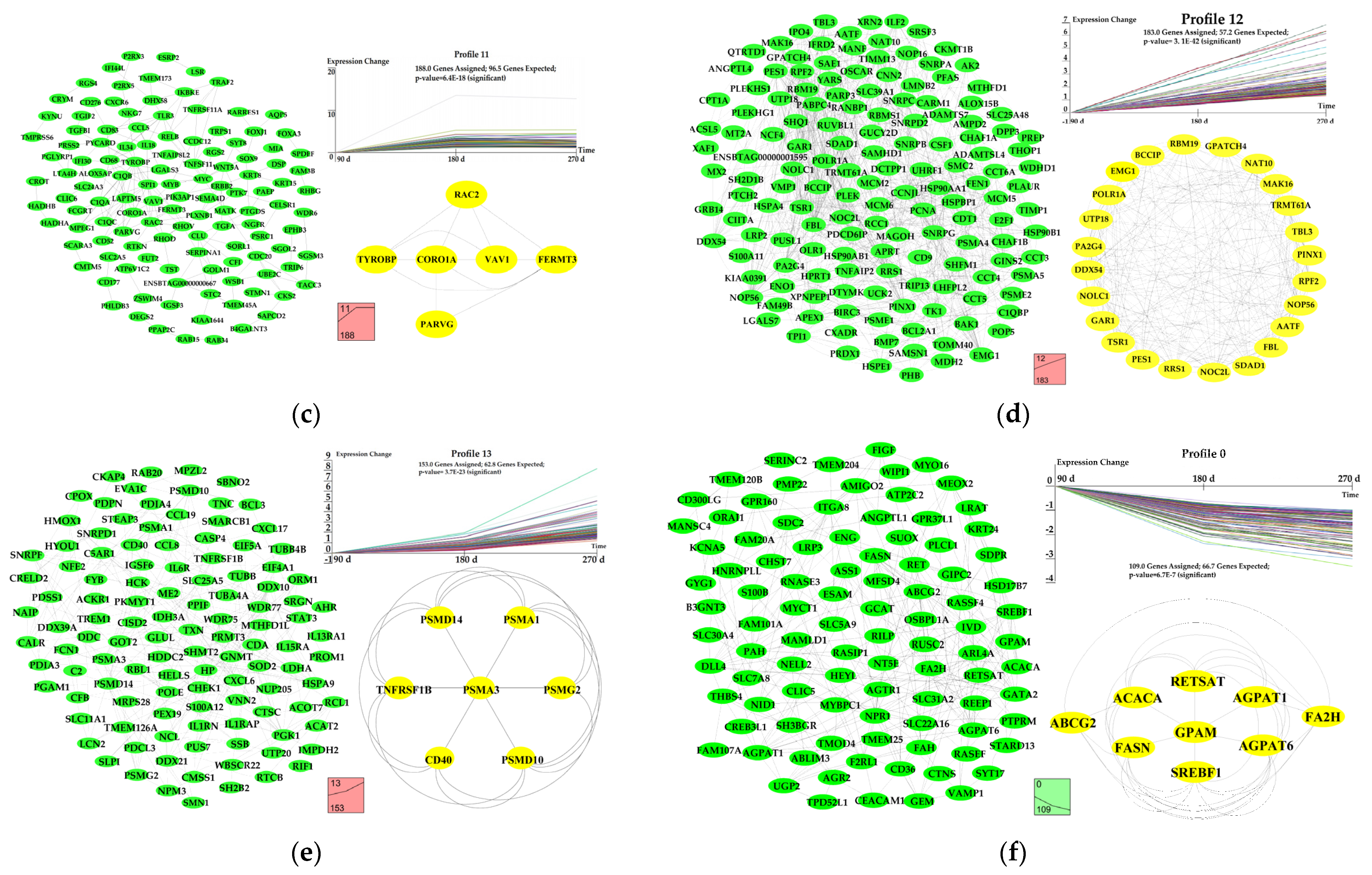
3.6. Dynamic Expression Profiles of DEGs
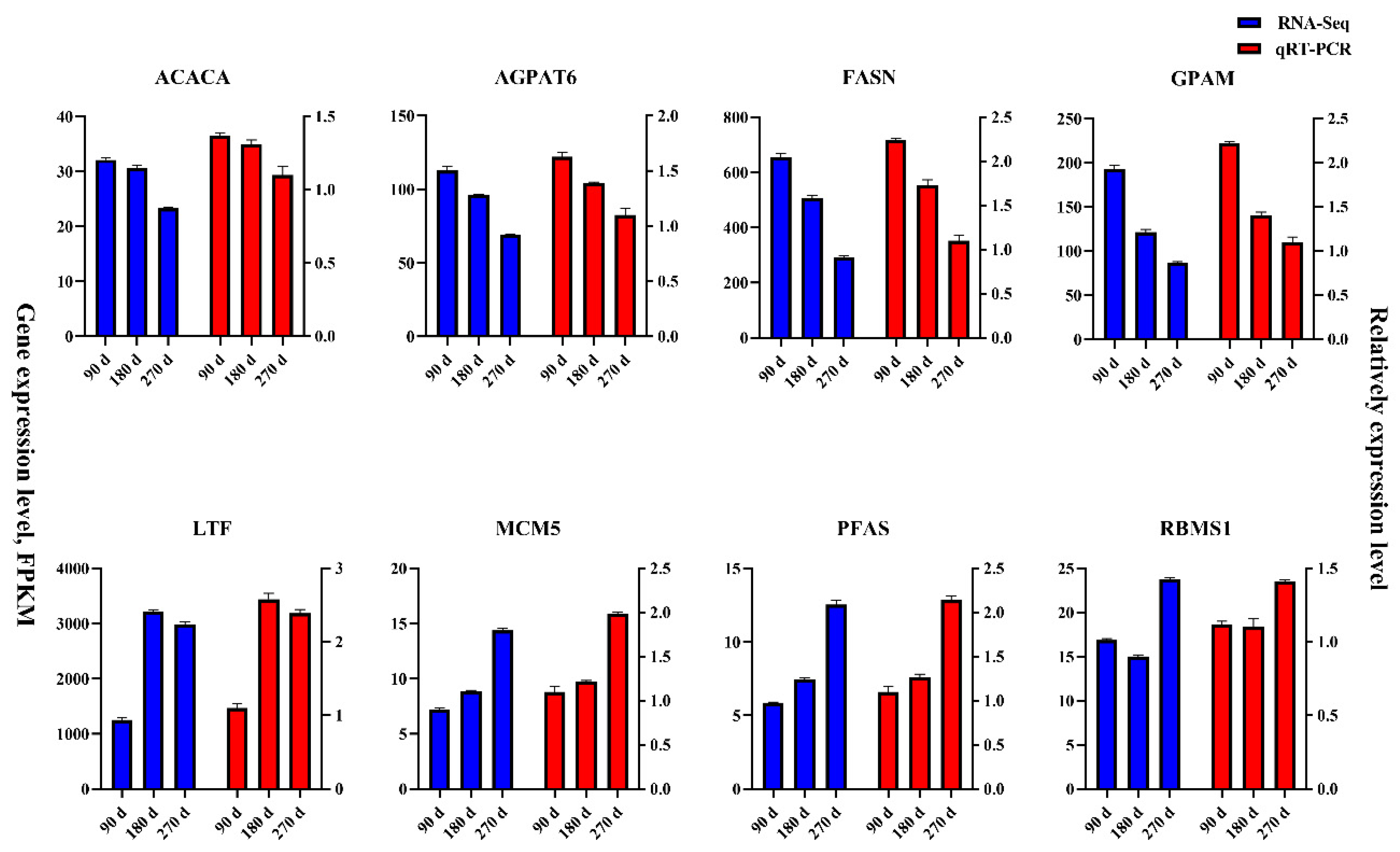
3.7. Verification Transcriptome Data of qRT-PCR
4. Discussion
4.1. Differentially Expressed Genes during Peak and Mid-Lactation
4.2. Differentially Expressed Genes during Peak and Late Lactation
4.3. Differentially Expressed Genes Related in Milk Fat Synthesis
4.4. Differentially Expressed Genes Related in Milk Protein Synthesis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Caroli, A.M.; Chessa, S.; Erhardt, G.J. Invited review: Milk protein polymorphisms in cattle: Effect on animal breeding and human nutrition. J. Dairy Sci. 2009, 92, 5335–5352. [Google Scholar] [CrossRef] [Green Version]
- Ji, Z.; Liu, Z.; Chao, T.; Hou, L.; Fan, R.; He, R.; Wang, G.; Wang, J. Screening of miRNA profiles and construction of regulation networks in early and late lactation of dairy goat mammary glands. Sci. Rep. 2017, 7, 11933. [Google Scholar] [CrossRef] [PubMed]
- Jeong, W.; Bae, H.; Lim, W.; Bazer, F.W.; Lee, H.; Song, G. The functional effects and mechanisms by which fibroblast growth factor 2 (FGF2) controls bovine mammary epithelial cells: Implications for the development and functionality of the bovine mammary gland. J. Anim. Sci. 2017, 95, 5365–5377. [Google Scholar] [CrossRef] [PubMed]
- Arora, R.; Sharma, A.; Sharma, U.; Girdhar, Y.; Kaur, M.; Kapoor, P.; Ahlawat, S.; Vijh, R.K. Buffalo milk transcriptome: A comparative analysis of early, mid and late lactation. Sci. Rep. 2019, 9, 5993. [Google Scholar] [CrossRef] [Green Version]
- Smit, L.E.; Schönfeldt, H.C.; Beer, W.H.J.D.; Smith, M.F. The Effect of Locality and Season on the Composition of South African Whole Milk. J. Food Compos. Anal. 2000, 13, 345–367. [Google Scholar] [CrossRef]
- Vijayakumar, M.; Park, J.H.; Ki, K.S.; Lim, D.H.; Kim, S.B.; Park, S.M.; Jeong, H.Y.; Park, B.Y.; Kim, T.I. The effect of lactation number, stage, length, and milking frequency on milk yield in Korean Holstein dairy cows using automatic milking system. Asian-Australas J. Anim. Sci. 2017, 30, 1093–1098. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.T.; Zou, Y.X.; White, R.R.; Liu, J.X.; Liu, H.Y. Transcriptomic profiles of the bovine mammary gland during lactation and the dry period. Funct. Integr. Genom. 2018, 18, 125–140. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Lv, H.; Jiang, M.; Zhou, J.; Song, S.; Hou, X. Functional analysis of the dairy cow mammary transcriptome between early lactation and mid-dry period. J. Dairy Res. 2019, 86, 63–67. [Google Scholar] [CrossRef]
- Suárez-Vega, A.; Gutiérrez-Gil, B.; Klopp, C.; Robert-Granie, C.; Tosser-Klopp, G.; Arranz, J.J. Characterization and Comparative Analysis of the Milk Transcriptome in Two Dairy Sheep Breeds using RNA Sequencing. Sci. Rep. 2015, 5, 18399. [Google Scholar] [CrossRef] [Green Version]
- Bionaz, M.; Periasamy, K.; Rodriguez-Zas, S.L.; Everts, R.E.; Lewin, H.A.; Hurley, W.L.; Loor, J.J. Old and new stories: Revelations from functional analysis of the bovine mammary transcriptome during the lactation cycle. PLoS ONE 2012, 7, e33268. [Google Scholar] [CrossRef]
- Zhao, S.; Fung-Leung, W.P.; Bittner, A.; Ngo, K.; Liu, X. Comparison of RNA-Seq and microarray in transcriptome profiling of activated T cells. PLoS ONE 2014, 9, e78644. [Google Scholar] [CrossRef]
- Gao, Y.; Lin, X.; Shi, K.; Yan, Z.; Wang, Z. Bovine mammary gene expression profiling during the onset of lactation. PLoS ONE 2013, 8, e70393. [Google Scholar] [CrossRef] [Green Version]
- Pizzi, C.; Bortoluzzi, S.; Bisognin, A.; Coppe, A.; Danieli, G.A. Detecting seeded motifs in DNA sequences. Nucleic Acids Res. 2005, 33, e135. [Google Scholar] [CrossRef]
- Ernst, J.; Bar-Joseph, Z. STEM: A tool for the analysis of short time series gene expression data. BMC Bioinform. 2006, 7, 191. [Google Scholar] [CrossRef] [Green Version]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Fan, W.; Mao, Y.; Yang, Z.; Lu, G.; Zhang, R.; Zhang, H.; Szeto, C.; Wang, C. Bovine leukemia virus infection in cattle of China: Association with reduced milk production and increased somatic cell score. J. Dairy Sci. 2016, 99, 3688–3697. [Google Scholar] [CrossRef] [Green Version]
- Piantoni, P.; Wang, P.; Drackley, J.K.; Hurley, W.L.; Loor, J.J. Expression of metabolic, tissue remodeling, oxidative stress, and inflammatory pathways in mammary tissue during involution in lactating dairy cows. Bioinform. Biol. Insights 2010, 4, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Cai, W.; Zhou, C.; Yin, H.; Zhang, Z.; Loor, J.J.; Sun, D.; Zhang, Q.; Liu, J.; Zhang, S. RNA-Seq reveals 10 novel promising candidate genes affecting milk protein concentration in the Chinese Holstein population. Sci. Rep. 2016, 6, 26813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Yang, M.; Cai, D.; Hao, Y.; Zhao, X.; Zhu, Y.; Zhu, H.; Yang, Z. Composition, coagulation characteristics, and cheese making capacity of yak milk. J. Dairy Sci. 2020, 103, 1276–1288. [Google Scholar] [CrossRef]
- Li, R.; Zhang, C.L.; Liao, X.X.; Chen, D.; Wang, W.Q.; Zhu, Y.H.; Geng, X.H.; Ji, D.J.; Mao, Y.J.; Gong, Y.C.; et al. Transcriptome microRNA profiling of bovine mammary glands infected with Staphylococcus aureus. Int. J. Mol. Sci. 2015, 16, 4997–5013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, B.; Yao, C.; Wang, Y.; Mao, S.; Wang, Y.; Wu, R.; Feng, W.; Chen, Y.; Yang, J.; Xue, C.; et al. The Landscape of Gene Expression and Molecular Regulation Following Spinal Cord Hemisection in Rats. Front. Mol. Neurosci. 2019, 12, 287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, L.; Sahana, G.; Su, G.; Yu, Y.; Zhang, S.; Lund, M.S.; Sørensen, P. Integrating Sequence-based GWAS and RNA-Seq Provides Novel Insights into the Genetic Basis of Mastitis and Milk Production in Dairy Cattle. Sci. Rep. 2017, 7, 45560. [Google Scholar] [CrossRef] [PubMed]
- Hurley, D.; Luque-Sastre, L.; Parker, C.T.; Huynh, S.; Eshwar, A.K.; Nguyen, S.V.; Andrews, N.; Moura, A.; Fox, E.M.; Jordan, K.; et al. Whole-Genome Sequencing-Based Characterization of 100 Listeria monocytogenes Isolates Collected from Food Processing Environments over a Four-Year Period. mSphere 2019, 4. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Wu, P.; Shen, M.; He, M.; Chen, L.; Qiu, C.; Shi, H.; Zhang, T.; Wang, J.; Xie, K.; et al. Transcriptome Analysis of Differentially Expressed Genes Related to the Growth and Development of the Jinghai Yellow Chicken. Genes 2019, 10, 539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kvist, J.; Gonçalves Athanàsio, C.; Shams Solari, O.; Brown, J.B.; Colbourne, J.K.; Pfrender, M.E.; Mirbahai, L. Pattern of DNA Methylation in Daphnia: Evolutionary Perspective. Genome Biol. Evol. 2018, 10, 1988–2007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, M.; Chatterjee, S.S.; Jain, S.; Katari, M.; DasGupta, R. TCF7L1 Modulates Colorectal Cancer Growth by Inhibiting Expression of the Tumor-Suppressor Gene EPHB3. Sci. Rep. 2016, 6, 28299. [Google Scholar] [CrossRef] [Green Version]
- Wen, D.; Xu, H.; Xie, L.; He, M.; Hou, H.; Wu, C.; Li, Y.; Zhang, C. Effects of Nitrogen Level during Seed Production on Wheat Seed Vigor and Seedling Establishment at the Transcriptome Level. Int. J. Mol. Sci. 2018, 19, 3417. [Google Scholar] [CrossRef] [Green Version]
- Tzin, V.; Hojo, Y.; Strickler, S.R.; Bartsch, L.J.; Archer, C.M.; Ahern, K.R.; Zhou, S.; Christensen, S.A.; Galis, I.; Mueller, L.A.; et al. Rapid defense responses in maize leaves induced by Spodoptera exigua caterpillar feeding. J. Exp. Bot. 2017, 68, 4709–4723. [Google Scholar] [CrossRef]
- Petty, L.E.; Highland, H.M.; Gamazon, E.R.; Hu, H.; Karhade, M.; Chen, H.H.; de Vries, P.S.; Grove, M.L.; Aguilar, D.; Bell, G.I.; et al. Functionally oriented analysis of cardiometabolic traits in a trans-ethnic sample. Hum. Mol. Genet. 2019, 28, 1212–1224. [Google Scholar] [CrossRef]
- Bian, J.; Deng, P.; Zhan, H.; Wu, X.; Nishantha, M.; Yan, Z.; Du, X.; Nie, X.; Song, W. Transcriptional Dynamics of Grain Development in Barley (Hordeum vulgare L.). Int. J. Mol. Sci. 2019, 20, 962. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.J.; Zhao, Q.Q.; Liu, X.S.; Dong, S.H.; E, J.F.; Li, X.; Liu, C.; Wang, H. Toll-like receptor 4 regulates spontaneous intestinal tumorigenesis by up-regulating IL-6 and GM-CSF. J. Cell Mol. Med. 2020, 24, 385–397. [Google Scholar] [CrossRef] [Green Version]
- Duarte-Delgado, D.; Dadshani, S.; Schoof, H.; Oyiga, B.C.; Schneider, M.; Mathew, B.; Léon, J.; Ballvora, A. Transcriptome profiling at osmotic and ionic phases of salt stress response in bread wheat uncovers trait-specific candidate genes. BMC Plant. Biol. 2020, 20, 428. [Google Scholar] [CrossRef]
- Ghandhi, S.A.; Sinha, A.; Markatou, M.; Amundson, S.A. Time-series clustering of gene expression in irradiated and bystander fibroblasts: An application of FBPA clustering. BMC Genom. 2011, 12, 2. [Google Scholar] [CrossRef] [Green Version]
- Kononikhin, A.S.; Brzhozovskiy, A.G.; Ryabokon, A.M.; Fedorchenko, K.; Zakharova, N.V.; Spasskii, A.I.; Popov, I.A.; Ilyin, V.K.; Solovyova, Z.O.; Pastushkova, L.K.; et al. Proteome Profiling of the Exhaled Breath Condensate after Long-Term Spaceflights. Int. J. Mol. Sci. 2019, 20, 4518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marburger, S.; Monnahan, P.; Seear, P.J.; Martin, S.H.; Koch, J.; Paajanen, P.; Bohutínská, M.; Higgins, J.D.; Schmickl, R.; Yant, L. Interspecific introgression mediates adaptation to whole genome duplication. Nat. Commun. 2019, 10, 5218. [Google Scholar] [CrossRef] [Green Version]
- Bionaz, M.; Loor, J.J. Identification of reference genes for quantitative real-time PCR in the bovine mammary gland during the lactation cycle. Physiol. Genom. 2007, 29, 312–319. [Google Scholar] [CrossRef] [PubMed]
- Grubišić, V.; McClain, J.L.; Fried, D.E.; Grants, I.; Rajasekhar, P.; Csizmadia, E.; Ajijola, O.A.; Watson, R.E.; Poole, D.P.; Robson, S.C.; et al. Enteric Glia Modulate Macrophage Phenotype and Visceral Sensitivity following Inflammation. Cell Rep. 2020, 32, 108100. [Google Scholar] [CrossRef] [PubMed]
- Capuco, A.V.; Wood, D.L.; Baldwin, R.; McLeod, K.; Paape, M.J. Mammary cell number, proliferation, and apoptosis during a bovine lactation: Relation to milk production and effect of bST. J. Dairy Sci. 2001, 84, 2177–2187. [Google Scholar] [CrossRef]
- Ollier, S.; Robert-Granié, C.; Bernard, L.; Chilliard, Y.; Leroux, C. Mammary transcriptome analysis of food-deprived lactating goats highlights genes involved in milk secretion and programmed cell death. J. Nutr. 2007, 137, 560–567. [Google Scholar] [CrossRef] [Green Version]
- Bionaz, M.; Loor, J.J. Gene networks driving bovine milk fat synthesis during the lactation cycle. BMC Genom. 2008, 9, 366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morammazi, S.; Masoudi, A.A.; Vaez Torshizi, R.; Pakdel, A. Differential Expression of the Alpha S1 Casein and Beta-Lactoglobulin Genes in Different Physiological Stages of the Adani Goats Mammary Glands. Iran. J. Biotechnol. 2016, 14, 278–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Fakharany, E.M.; El-Baky, N.A.; Linjawi, M.H.; Aljaddawi, A.A.; Saleem, T.H.; Nassar, A.Y.; Osman, A.; Redwan, E.M. Influence of camel milk on the hepatitis C virus burden of infected patients. Exp. Ther. Med. 2017, 13, 1313–1320. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Hu, X.; Long, K.; Gao, C.; Dong, H.L.; Zhong, Q.; Gao, X.M.; Gong, F.Y. Extraordinarily potent proinflammatory properties of lactoferrin-containing immunocomplexes against human monocytes and macrophages. Sci. Rep. 2017, 7, 4230. [Google Scholar] [CrossRef]
- Genet, F.; Schnitzler, A.; Droz-Bartholet, F.; Salga, M.; Tatu, L.; Debaud, C.; Denormandie, P.; Parratte, B. Successive motor nerve blocks to identify the muscles causing a spasticity pattern: Example of the arm flexion pattern. J. Anat. 2017, 230, 106–116. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Yue, T.; Ahmad, M.J.; Hu, X.; Zhang, X.; Deng, T.; Hu, Y.; He, C.; Zhou, Y.; Yang, L. Transcriptome Analysis Reveals Potential Regulatory Genes Related to Heat Tolerance in Holstein Dairy Cattle. Genes 2020, 11, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, S.; Singh, P.; Sharma, A.; Arora, S.; Shriwash, N.; Rahmani, A.H.; Almatroodi, S.A.; Manda, K.; Dohare, R.; Syed, M.A. Transcriptome Meta-Analysis Deciphers a Dysregulation in Immune Response-Associated Gene Signatures during Sepsis. Genes 2019, 10, 5. [Google Scholar] [CrossRef] [Green Version]
- Jin, F.; Wang, K.; Sun, X.; Zhang, Z.; Han, P. Gene expression analysis: Regulation of key genes associated with mycophenolate mofetil treatment of symptomatic carotid artery stenosis. Mol. Med. Rep. 2017, 16, 7450–7458. [Google Scholar] [CrossRef] [Green Version]
- Moisá, S.J.; Ji, P.; Drackley, J.K.; Rodriguez-Zas, S.L.; Loor, J.J. Transcriptional changes in mesenteric and subcutaneous adipose tissue from Holstein cows in response to plane of dietary energy. J. Anim. Sci. Biotechnol. 2017, 8, 85. [Google Scholar] [CrossRef] [PubMed]
- Antony-Debré, I.; Manchev, V.T.; Balayn, N.; Bluteau, D.; Tomowiak, C.; Legrand, C.; Langlois, T.; Bawa, O.; Tosca, L.; Tachdjian, G.; et al. Level of RUNX1 activity is critical for leukemic predisposition but not for thrombocytopenia. Blood 2015, 125, 930–940. [Google Scholar] [CrossRef] [Green Version]
- Vangelisti, A.; Zambrano, L.S.; Caruso, G.; Macheda, D.; Bernardi, R.; Usai, G.; Mascagni, F.; Giordani, T.; Gucci, R.; Cavallini, A.; et al. How an ancient, salt-tolerant fruit crop, Ficus carica L., copes with salinity: A transcriptome analysis. Sci. Rep. 2019, 9, 2561. [Google Scholar] [CrossRef] [Green Version]
- Gu, J.; Zeng, Z.; Wang, Y.; Lyu, Y. Transcriptome Analysis of Carbohydrate Metabolism Genes and Molecular Regulation of Sucrose Transport Gene LoSUT on the Flowering Process of Developing Oriental Hybrid Lily ‘Sorbonne’ Bulb. Int. J. Mol. Sci. 2020, 21, 3092. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Yong, Y.; Zhang, Y.; Lyu, Y. Transcriptional Regulatory Network of GA Floral Induction Pathway in LA Hybrid Lily. Int J. Mol. Sci. 2019, 20, 2694. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Jose, C.; Kenney, N.; Morrison, B.; Cutler, M.L. Global expression profiling reveals regulation of CTGF/CCN2 during lactogenic differentiation. J. Cell Commun. Signal. 2009, 3, 43–55. [Google Scholar] [CrossRef] [Green Version]
- Marti, A.; Lazar, H.; Ritter, P.; Jaggi, R. Transcription factor activities and gene expression during mouse mammary gland involution. J. Mammary Gland Biol. Neoplasia 1999, 4, 145–152. [Google Scholar] [CrossRef]
- Lacasse, P.; Lollivier, V.; Dessauge, F.; Bruckmaier, R.M.; Ollier, S.; Boutinaud, M. New developments on the galactopoietic role of prolactin in dairy ruminants. Domest. Anim. Endocrinol. 2012, 43, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Ollier, S.; Zhao, X.; Lacasse, P. Effects of feed restriction and prolactin-release inhibition at drying off on metabolism and mammary gland involution in cows. J. Dairy Sci. 2014, 97, 4942–4954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mach, N.; Jacobs, A.A.; Kruijt, L.; van Baal, J.; Smits, M.A. Alteration of gene expression in mammary gland tissue of dairy cows in response to dietary unsaturated fatty acids. Animal 2011, 5, 1217–1230. [Google Scholar] [CrossRef] [Green Version]
- Mach, N.; van Baal, J.; Kruijt, L.; Jacobs, A.; Smits, M. Dietary unsaturated fatty acids affect the mammary gland integrity and health in lactating dairy cows. BMC Proc. 2011, 5 (Suppl. 4), S35. [Google Scholar] [CrossRef]
- Lucena, R.; Gallego, M.; Cárdenas, S.; Valcárcel, M. Autoanalyzer for milk quality control based on the lactose, fat, and total protein contents. Anal. Chem. 2003, 75, 1425–1429. [Google Scholar] [CrossRef]
- Shingfield, K.J.; Chilliard, Y.; Toivonen, V.; Kairenius, P.; Givens, D.I. Trans fatty acids and bioactive lipids in ruminant milk. Adv. Exp. Med. Biol. 2008, 606, 3–65. [Google Scholar] [CrossRef]
- Angulo, J.; Mahecha, L.; Nuernberg, K.; Nuernberg, G.; Dannenberger, D.; Olivera, M.; Boutinaud, M.; Leroux, C.; Albrecht, E.; Bernard, L. Effects of polyunsaturated fatty acids from plant oils and algae on milk fat yield and composition are associated with mammary lipogenic and SREBF1 gene expression. Animal 2012, 6, 1961–1972. [Google Scholar] [CrossRef] [Green Version]
- Mattes, R.D. Oral detection of short-, medium-, and long-chain free fatty acids in humans. Chem. Senses 2009, 34, 145–150. [Google Scholar] [CrossRef] [Green Version]
- Hussain, S.A.; Hameed, A.; Khan, M.A.K.; Zhang, Y.; Zhang, H.; Garre, V.; Song, Y. Engineering of Fatty Acid Synthases (FASs) to Boost the Production of Medium-Chain Fatty Acids (MCFAs) in Mucor circinelloides. Int. J. Mol. Sci. 2019, 20, 786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanos, R.; Murray, I.A.; Smith, P.B.; Patterson, A.; Perdew, G.H. Role of the Ah receptor in homeostatic control of fatty acid synthesis in the liver. Toxicol. Sci. 2012, 129, 372–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, X.H.; Mao, J.H.; Peng, A.F.; Zhou, Y.; Huang, S.H.; Liu, Z.L. Tumor suppressive microRNA-424 inhibits osteosarcoma cell migration and invasion via targeting fatty acid synthase. Exp. Ther. Med. 2013, 5, 1048–1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, G.; Yu, X.; Wang, J. Network-aided Bi-Clustering for discovering cancer subtypes. Sci. Rep. 2017, 7, 1046. [Google Scholar] [CrossRef] [PubMed]
- Das, J.K.; Choudhury, P.P.; Chaudhuri, A.; Hassan, S.S.; Basu, P. Analysis of Purines and Pyrimidines distribution over miRNAs of Human, Gorilla, Chimpanzee, Mouse and Rat. Sci. Rep. 2018, 8, 9974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearsall, A.W.t.; Madanagopal, S.G.; Karas, S.G. Transfer of the latissimus dorsi as a salvage procedure for failed debridement and attempted repair of massive rotator cuff tears. Orthopedics 2007, 30, 943–949. [Google Scholar] [CrossRef] [PubMed]
- Dieni, C.A.; Storey, K.B. Regulation of 5′-adenosine monophosphate deaminase in the freeze tolerant wood frog, Rana sylvatica. BMC Biochem. 2008, 9, 12. [Google Scholar] [CrossRef] [Green Version]
- Gonçalves, V.; Henriques, A.F.; Pereira, J.F.; Neves Costa, A.; Moyer, M.P.; Moita, L.F.; Gama-Carvalho, M.; Matos, P.; Jordan, P. Phosphorylation of SRSF1 by SRPK1 regulates alternative splicing of tumor-related Rac1b in colorectal cells. Rna 2014, 20, 474–482. [Google Scholar] [CrossRef] [Green Version]
- Ohtani, M. Plant snRNP Biogenesis: A Perspective from the Nucleolus and Cajal Bodies. Front. Plant. Sci. 2017, 8, 2184. [Google Scholar] [CrossRef] [Green Version]
- Kuhle, B.; Ficner, R. eIF5B employs a novel domain release mechanism to catalyze ribosomal subunit joining. EMBO J. 2014, 33, 1177–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sardana, R.; White, J.P.; Johnson, A.W. The rRNA methyltransferase Bud23 shows functional interaction with components of the SSU processome and RNase MRP. RNA 2013, 19, 828–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, R.M.; Cai, L.; Zhang, W.P.; Cao, H.L.; Qian, P.Y. Rare Events of Intragenus and Intraspecies Horizontal Transfer of the 16S rRNA Gene. Genome Biol. Evol. 2015, 7, 2310–2320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitterer, V.; Shayan, R.; Ferreira-Cerca, S.; Murat, G.; Enne, T.; Rinaldi, D.; Weigl, S.; Omanic, H.; Gleizes, P.E.; Kressler, D.; et al. Conformational proofreading of distant 40S ribosomal subunit maturation events by a long-range communication mechanism. Nat. Commun. 2019, 10, 2754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Léger-Silvestre, I.; Milkereit, P.; Ferreira-Cerca, S.; Saveanu, C.; Rousselle, J.C.; Choesmel, V.; Guinefoleau, C.; Gas, N.; Gleizes, P.E. The ribosomal protein Rps15p is required for nuclear exit of the 40S subunit precursors in yeast. EMBO J. 2004, 23, 2336–2347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, R.; Wang, M.; Zhou, J.; Fu, Y.; Liang, M.; Guo, D.; Nie, J. Prediction of Enzyme Function Based on Three Parallel Deep CNN and Amino Acid Mutation. Int. J. Mol. Sci. 2019, 20, 2845. [Google Scholar] [CrossRef] [Green Version]
- Tu, R.; Qian, J.; Rui, M.; Tao, N.; Sun, M.; Zhuang, Y.; Lv, H.; Han, J.; Li, M.; Xie, W. Proteolytic cleavage is required for functional neuroligin 2 maturation and trafficking in Drosophila. J. Mol. Cell Biol. 2017, 9, 231–242. [Google Scholar] [CrossRef] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lactation Days | 90 Days | 180 Days | 270 Days | p-Value |
|---|---|---|---|---|
| Milk yield (kg) | 34.52 ± 1.72 a | 31.23 ± 1.53 b | 26.37 ± 1.21 c | <0.05 |
| Milk protein (g/100 g) | 3.05 ± 0.07 c | 3.27 ± 0.05 b | 3.53 ± 0.06 a | <0.05 |
| Milk protein (g/day) | 1053.16 ± 29.25 | 1023.87 ± 17.39 | 931.27 ± 20.11 | >0.05 |
| Milk fat (g/100 g) | 3.26 ± 0.08 c | 3.61 ± 0.07 b | 3.99 ± 0.08 a | <0.05 |
| Milk fat (g/day) | 1124.96 ± 28.63 | 1126.79 ± 26.18 | 1053.36 ± 20.46 | >0.05 |
| Lactose (g/100 g) | 5.24 ± 0.10 a | 4.98 ± 0.09 b | 4.70 ± 0.07 c | <0.05 |
| Lactose (g/day) | 1809.13 ± 42.17 | 1556.46 ± 22.65 | 1240.23 ± 28.62 | <0.05 |
| Somatic cell count (SCC) (104/mL) | 23.82 | 32.57 | 47.22 | - |
| Somatic cell score (SCS) | 4.25 ± 0.02 c | 4.70 ± 0.03 b | 5.24 ± 0.02 a | <0.05 |
| Lactation Days | 90 Days | 180 Days | 270 Days | p-Value |
|---|---|---|---|---|
| Average acinus area (μm2) | 378,109 ± 43,965 a | 154,808 ± 16,671 b | 28,361 ± 2107 c | <0.05 |
| The number of nuclei per acinus | 42.50 ± 2.12 a | 20.50 ± 2.12 b | 13.50 ± 0.71 c | <0.05 |
| The average area of lipid droplets (μm2) | 1348.73 ± 56.33 | 1336.74 ± 69.80 | 1355.03 ± 35.65 | >0.05 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fan, Y.; Han, Z.; Lu, X.; Arbab, A.A.I.; Nazar, M.; Yang, Y.; Yang, Z. Short Time-Series Expression Transcriptome Data Reveal the Gene Expression Patterns of Dairy Cow Mammary Gland as Milk Yield Decreased Process. Genes 2021, 12, 942. https://doi.org/10.3390/genes12060942
Fan Y, Han Z, Lu X, Arbab AAI, Nazar M, Yang Y, Yang Z. Short Time-Series Expression Transcriptome Data Reveal the Gene Expression Patterns of Dairy Cow Mammary Gland as Milk Yield Decreased Process. Genes. 2021; 12(6):942. https://doi.org/10.3390/genes12060942
Chicago/Turabian StyleFan, Yongliang, Ziyin Han, Xubin Lu, Abdelaziz Adam Idriss Arbab, Mudasir Nazar, Yi Yang, and Zhangping Yang. 2021. "Short Time-Series Expression Transcriptome Data Reveal the Gene Expression Patterns of Dairy Cow Mammary Gland as Milk Yield Decreased Process" Genes 12, no. 6: 942. https://doi.org/10.3390/genes12060942
APA StyleFan, Y., Han, Z., Lu, X., Arbab, A. A. I., Nazar, M., Yang, Y., & Yang, Z. (2021). Short Time-Series Expression Transcriptome Data Reveal the Gene Expression Patterns of Dairy Cow Mammary Gland as Milk Yield Decreased Process. Genes, 12(6), 942. https://doi.org/10.3390/genes12060942