
QTL Analysis of Stem Elongation and Flowering Time in Lettuce Using Genotyping-by-Sequencing


Abstract
:1. Introduction
2. Material and Methods
2.1. Mapping Population
2.2. Phenotyping of the RIL Population
2.3. Genotyping-by-Sequencing
2.4. Linkage Map Construction and QTL Mapping
2.5. Whole Genome Resequencing and Annotation
3. Results
3.1. Trait Variation
3.2. Correlations between Traits
3.3. SNP Discovery by GBS and Construction of Genetic Map
3.4. QTL Analysis
3.5. Candidate Gene Prediction for QTLs Controlling Stem Elongation and Bolting
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chen, Z.; Han, Y.; Ning, K.; Ding, Y.; Zhao, W.; Yan, S.; Luo, C.; Jiang, X.; Ge, D.; Liu, R. Inflorescence development and the role of LsFT in regulating bolting in lettuce (Lactuca sativa L.). Front. Plant Sci. 2018, 8, 2248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuda, M.; Matsuo, S.; Kikuchi, K.; Kawazu, Y.; Fujiyama, R.; Honda, I. Isolation and functional characterization of the FLOWERING LOCUS T homolog, the LsFT gene, in lettuce. J. Plant Physiol. 2011, 168, 1602–1607. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Chin-Wo, S.; Wang, Z.; Yang, X.; Kozik, A.; Arikit, S.; Song, C.; Xia, L.; Froenicke, L.; Lavelle, D.O.; Truco, M.-J. Genome assembly with in vitro proximity ligation data and whole-genome triplication in lettuce. Nat. Commun. 2017, 8, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Hooper, L.; Cassidy, A. A review of the health care potential of bioactive compounds. J. Sci. Food Agric. 2006, 86, 1805–1813. [Google Scholar] [CrossRef]
- Ito, H.; Kato, T.; Konno, Y. Factors associated with the flower induction in lettuce. Tohoku J. Agric. Res. 1963, 14, 51–65. [Google Scholar]
- Lee, O.N.; Sugiyama, N.; Kosuge, S. Allometry of stem growth in lettuce plants. J. Jpn. Soc. Hortic. Sci. 2003, 72, 24–28. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Huang, W.; Hou, K.; Wu, W. Bolting, an important process in plant development, two types in plants. J. Plant Biol. 2019, 62, 161–169. [Google Scholar] [CrossRef]
- Alvarez-Buylla, E.R.; Benítez, M.; Corvera-Poiré, A.; Cador, Á.C.; de Folter, S.; de Buen, A.G.; Garay-Arroyo, A.; García-Ponce, B.; Jaimes-Miranda, F.; Pérez-Ruiz, R.V. Flower development. Am. Soc. Plant Biol. 2010, 8, e0127. [Google Scholar] [CrossRef] [Green Version]
- Lee, O.N.; Nemoto, K.; Sugiyama, N. Histone H4 gene expression in shoot apices associated with floral initiation in lettuce. J. Jpn. Soc. Hortic. Sci. 2005, 74, 121–126. [Google Scholar] [CrossRef] [Green Version]
- Ryder, E.J.; Milligan, D.C. Additional genes controlling flowering time in Lactuca sativa and L. serriola. J. Jpn. Soc. Hortic. Sci. 2005, 130, 448–453. [Google Scholar] [CrossRef] [Green Version]
- Johnson, W.; Jackson, L.; Ochoa, O.; Van Wijk, R.; Peleman, J.; Clair, D.S.; Michelmore, R.W. Lettuce, a shallow-rooted crop, and Lactuca serriola, its wild progenitor, differ at QTL determining root architecture and deep soil water exploitation. Theor. Appl. Genet. 2000, 101, 1066–1073. [Google Scholar] [CrossRef]
- Poland, J.A.; Brown, P.J.; Sorrells, M.E.; Jannink, J.-L. Development of high-density genetic maps for barley and wheat using a novel two-enzyme genotyping-by-sequencing approach. PLoS ONE 2012, 7, e32253. [Google Scholar] [CrossRef] [Green Version]
- Argyris, J.; Truco, M.J.; Ochoa, O.; Knapp, S.J.; Still, D.W.; Lenssen, G.M.; Schut, J.W.; Michelmore, R.W.; Bradford, K.J. Quantitative trait loci associated with seed and seedling traits in Lactuca. Theor. Appl. Genet. 2005, 111, 1365–1376. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.Z.; Wagstaff, C.; Rae, A.M.; Sihota, A.K.; Keevil, C.W.; Rothwell, S.D.; Clarkson, G.J.; Michelmore, R.W.; Truco, M.J.; Dixon, M.S. QTLs for shelf life in lettuce co-locate with those for leaf biophysical properties but not with those for leaf developmental traits. J. Exp. Bot. 2007, 58, 1433–1449. [Google Scholar] [CrossRef] [Green Version]
- Jeuken, M.; Lindhout, P. Lactuca saligna, a non-host for lettuce downy mildew (Bremia lactucae), harbors a new race-specific Dm gene and three QTLs for resistance. Theor. Appl. Genet. 2002, 105, 384–391. [Google Scholar] [CrossRef] [PubMed]
- Mamo, B.E.; Hayes, R.J.; Truco, M.J.; Puri, K.D.; Michelmore, R.W.; Subbarao, K.V.; Simko, I. The genetics of resistance to lettuce drop (Sclerotinia spp.) in lettuce in a recombinant inbred line population from Reine des Glaces× Eruption. Theor. Appl. Genet. 2019, 132, 2439–2460. [Google Scholar] [CrossRef] [PubMed]
- Jenni, S.; Truco, M.J.; Michelmore, R.W. Quantitative trait loci associated with tipburn, heat stress-induced physiological disorders, and maturity traits in crisphead lettuce. Theor. Appl. Genet. 2013, 126, 3065–3079. [Google Scholar] [CrossRef]
- Hartman, Y.; Hooftman, D.A.; Schranz, M.E.; van Tienderen, P.H. QTL analysis reveals the genetic architecture of domestication traits in Crisphead lettuce. Genet. Resour. Crop. Evol. 2013, 60, 1487–1500. [Google Scholar] [CrossRef] [Green Version]
- Hackett, C.A.; McLean, K.; Bryan, G.J. Linkage analysis and QTL mapping using SNP dosage data in a tetraploid potato mapping population. PLoS ONE 2013, 8, e63939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crossa, J.; Beyene, Y.; Kassa, S.; Pérez, P.; Hickey, J.M.; Chen, C.; de los Campos, G.; Burgueño, J.; Windhausen, V.S.; Buckler, E. Genomic prediction in maize breeding populations with genotyping-by-sequencing. G3 Genes Genom. Genet. 2013, 3, 1903–1926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gardner, K.M.; Brown, P.; Cooke, T.F.; Cann, S.; Costa, F.; Bustamante, C.; Velasco, R.; Troggio, M.; Myles, S. Fast and cost-effective genetic mapping in apple using next-generation sequencing. G3 Genes Genom. Genet. 2014, 4, 1681–1687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Bayer, M.; Druka, A.; Russell, J.R.; Hackett, C.A.; Poland, J.; Ramsay, L.; Hedley, P.E.; Waugh, R. An evaluation of genotyping by sequencing (GBS) to map the Breviaristatum-e (ari-e) locus in cultivated barley. BMC Genom. 2014, 15, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Sonah, H.; Bastien, M.; Iquira, E.; Tardivel, A.; Légaré, G.; Boyle, B.; Normandeau, É.; Laroche, J.; Larose, S.; Jean, M. An improved genotyping by sequencing (GBS) approach offering increased versatility and efficiency of SNP discovery and genotyping. PLoS ONE 2013, 8, e54603. [Google Scholar] [CrossRef] [Green Version]
- Spindel, J.; Wright, M.; Chen, C.; Cobb, J.; Gage, J.; Harrington, S.; Lorieux, M.; Ahmadi, N.; McCouch, S. Bridging the genotyping gap: Using genotyping by sequencing (GBS) to add high-density SNP markers and new value to traditional bi-parental mapping and breeding populations. Theor. Appl. Genet. 2013, 126, 2699–2716. [Google Scholar] [CrossRef] [Green Version]
- Verma, S.; Gupta, S.; Bandhiwal, N.; Kumar, T.; Bharadwaj, C.; Bhatia, S. High-density linkage map construction and mapping of seed trait QTLs in chickpea (Cicer arietinum L.) using Genotyping-by-Sequencing (GBS). Sci. Rep. 2015, 5, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef] [Green Version]
- Appleby, N.; Edwards, D.; Batley, J. New technologies for ultra-high throughput genotyping in plants. Plant Genom. 2009, 513, 19–39. [Google Scholar]
- Mammadov, J.; Aggarwal, R.; Buyyarapu, R.; Kumpatla, S. SNP markers and their impact on plant breeding. Int. J. Plant Genom. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- De Donato, M.; Peters, S.O.; Mitchell, S.E.; Hussain, T.; Imumorin, I.G. Genotyping-by-sequencing (GBS): A novel, efficient and cost-effective genotyping method for cattle using next-generation sequencing. PLoS ONE 2013, 8, e62137. [Google Scholar] [CrossRef]
- Catchen, J.; Hohenlohe, P.A.; Bassham, S.; Amores, A.; Cresko, W.A. Stacks: An analysis tool set for population genomics. Mol. Ecol. 2013, 22, 3124–3140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, J.; Butler, D.; Taylor, M.J. Package ‘ASMap’. 2018. Available online: http://bioconductor.statistik.tu-dortmund.de/cran/web/packages/ASMap/ASMap.pdf (accessed on 1 April 2018).
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data; Babraham Bioinformatics: Cambridge, UK, 2010. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M. The genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Truong, H.T.; Ramos, A.M.; Yalcin, F.; de Ruiter, M.; van der Poel, H.J.; Huvenaars, K.H.; Hogers, R.C.; van Enckevort, L.J.; Janssen, A.; van Orsouw, N.J. Sequence-based genotyping for marker discovery and co-dominant scoring in germplasm and populations. PLoS ONE 2012, 7, e37565. [Google Scholar] [CrossRef] [Green Version]
- He, J.; Zhao, X.; Laroche, A.; Lu, Z.-X.; Liu, H.; Li, Z. Genotyping-by-sequencing (GBS), an ultimate marker-assisted selection (MAS) tool to accelerate plant breeding. Front. Plant. Sci. 2014, 5. [Google Scholar] [CrossRef] [Green Version]
- Helentjaris, T.; Slocum, M.; Wright, S.; Schaefer, A.; Nienhuis, J. Construction of genetic linkage maps in maize and tomato using restriction fragment length polymorphisms. Theor. Appl. Genet. 1986, 72, 761–769. [Google Scholar] [CrossRef]
- Wickland, D.P.; Battu, G.; Hudson, K.A.; Diers, B.W.; Hudson, M.E. A comparison of genotyping-by-sequencing analysis methods on low-coverage crop datasets shows advantages of a new workflow, GB-eaSy. BMC Bioinform. 2017, 18, 1–12. [Google Scholar] [CrossRef]
- Damerval, C.; Maurice, A.; Josse, J.; De Vienne, D. Quantitative trait loci underlying gene product variation: A novel perspective for analyzing regulation of genome expression. Genetics 1994, 137, 289–301. [Google Scholar] [CrossRef]
- Flint-Garcia, S.A.; Thuillet, A.C.; Yu, J.; Pressoir, G.; Romero, S.M.; Mitchell, S.E.; Doebley, J.; Kresovich, S.; Goodman, M.M.; Buckler, E.S. Maize association population: A high-resolution platform for quantitative trait locus dissection. Plant J. 2005, 44, 1054–1064. [Google Scholar] [CrossRef]
- Sharbel, T.F.; Haubold, B.; Mitchell-Olds, T. Genetic isolation by distance in Arabidopsis thaliana: Biogeography and postglacial colonization of Europe. Mol. Ecol. 2000, 9, 2109–2118. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Gore, M.; Buckler, E.S.; Yu, J. Status and prospects of association mapping in plants. Plant Genome 2008, 1. [Google Scholar] [CrossRef]
- Deschamps, S.; Llaca, V.; May, G.D. Genotyping-by-sequencing in plants. Biology 2012, 1, 460–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, R.; Truco, M.J.; Lavelle, D.O.; Michelmore, R.W. A composite analysis of flowering time regulation in lettuce. Front. Plant Sci. 2021, 12, 360. [Google Scholar] [CrossRef]
- Park, S.; Kumar, P.; Shi, A.; Mou, B. Population genetics and genome-wide association studies provide insights into the influence of selective breeding on genetic variation in lettuce. Plant Genome 2021, e20086. [Google Scholar] [CrossRef]
- Sumugat, M.R.; Lee, O.N.; Nemoto, K.; Sugiyama, N. Quantitative trait loci analysis of flowering-time-related traits in tomato. Sci. Hortic. 2010, 123, 343–349. [Google Scholar] [CrossRef]
- Fan, C.; Yu, X.; Xing, Y.; Xu, C.; Luo, L.; Zhang, Q. The main effects, epistatic effects and environmental interactions of QTLs on the cooking and eating quality of rice in a doubled-haploid line population. Theor. Appl. Genet. 2005, 110, 1445–1452. [Google Scholar] [CrossRef] [PubMed]
- Burger, J.C.; Chapman, M.A.; Burke, J.M. Molecular insights into the evolution of crop plants. Am. J. Bot. 2008, 95, 113–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross-Ibarra, J. Quantitative trait loci and the study of plant domestication. Genet. Adap. 2005, 123, 197–204. [Google Scholar]
- Paterson, A.H.; Damon, S.; Hewitt, J.D.; Zamir, D.; Rabinowitch, H.D.; Lincoln, S.E.; Lander, E.S.; Tanksley, S.D. Mendelian factors underlying quantitative traits in tomato: Comparison across species, generations, and environments. Genetics 1991, 127, 181–197. [Google Scholar] [CrossRef] [PubMed]
- Basunanda, P.; Radoev, M.; Ecke, W.; Friedt, W.; Becker, H.; Snowdon, R. Comparative mapping of quantitative trait loci involved in heterosis for seedling and yield traits in oilseed rape (Brassica napus L.). Theor. Appl. Genet. 2010, 120, 271–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramchiary, N.; Padmaja, K.; Sharma, S.; Gupta, V.; Sodhi, Y.; Mukhopadhyay, A.; Arumugam, N.; Pental, D.; Pradhan, A. Mapping of yield influencing QTL in Brassica juncea: Implications for breeding of a major oilseed crop of dryland areas. Theor. Appl. Genet. 2007, 115, 807–817. [Google Scholar] [CrossRef] [PubMed]
- Koinange, E.M.; Singh, S.P.; Gepts, P. Genetic control of the domestication syndrome in common bean. Crop Sci. 1996, 36, 1037–1045. [Google Scholar] [CrossRef] [Green Version]
- Chiang, G.C.; Barua, D.; Kramer, E.M.; Amasino, R.M.; Donohue, K. Major flowering time gene, flowering locus C, regulates seed germination in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2009, 106, 11661–11666. [Google Scholar] [CrossRef] [Green Version]
- Woods, D.P.; Ream, T.S.; Minevich, G.; Hobert, O.; Amasino, R.M. PHYTOCHROME C is an essential light receptor for photoperiodic flowering in the temperate grass, Brachypodium distachyon. Genetics 2014, 198, 397–408. [Google Scholar] [CrossRef] [Green Version]
- Takano, M.; Inagaki, N.; Xie, X.; Yuzurihara, N.; Hihara, F.; Ishizuka, T.; Yano, M.; Nishimura, M.; Miyao, A.; Hirochika, H. Distinct and cooperative functions of phytochromes A, B, and C in the control of deetiolation and flowering in rice. Plant Cell 2005, 17, 3311–3325. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Wu, G.; Zhao, Y.; Wang, B.; Zhao, B.; Kong, D.; Wei, H.; Chen, C.; Wang, H. CRISPR/Cas9-mediated knockout and overexpression studies reveal a role of maize phytochrome C in regulating flowering time and plant height. Plant Biotechnol. J. 2020, 18, 2520–2532. [Google Scholar] [CrossRef]
- Lu, Y.; Yao, N.; Liu, X.; Chen, N.; Liu, H. Progress in studies of ZW10, a proper chromosome segregation protein. Biochem. Suppl. Ser. A Membr. Cell Biol. 2008, 2, 96–109. [Google Scholar] [CrossRef]
- Starr, D.A.; Williams, B.C.; Li, Z.; Etemad-Moghadam, B.; Dawe, R.K.; Goldberg, M.L. Conservation of the centromere/kinetochore protein ZW10. J. Cell Biol. 1997, 138, 1289–1301. [Google Scholar] [CrossRef] [PubMed]
- Morris, E.R.; Walker, J.C. Receptor-like protein kinases: The keys to response. Curr. Opin. Plant Biol. 2003, 6, 339–342. [Google Scholar] [CrossRef]
- Bian, Z.; Cheng, R.; Wang, Y.; Yang, Q.; Lu, C. Effect of green light on nitrate reduction and edible quality of hydroponically grown lettuce (Lactuca sativa L.) under short-term continuous light from red and blue light-emitting diodes. Environ. Exp. Bot. 2018, 153, 63–71. [Google Scholar] [CrossRef] [Green Version]
- Sivasankar, S.; Rothstein, S.; Oaks, A. Regulation of the accumulation and reduction of nitrate by nitrogen and carbon metabolites in maize seedlings. Plant Physiol. 1997, 114, 583–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manna, S. An overview of pentatricopeptide repeat proteins and their applications. Biochimie 2015, 113, 93–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
 ) F5 (n = 127).
) F5 (n = 127).
) F5 (n = 127).
) F5 (n = 127).


{kind=link}
{kind=link}
| Year | Trait | Chimasanchu (P1) | Banchu Red Fire (P2) | F5 |
|---|---|---|---|---|
| 2013 | DTB | 88.00 ± 6.24 | 86.80 ± 2.59 | 83.10 ± 14.43 |
| DTF | 111.60 ± 4.98 | 109.20 ± 3.90 | 105.48 ± 15.62 | |
| DTS | 124.20 ± 4.55 | 122.80 ± 3.90 | 118.69 ± 15.97 | |
| LN | 67.80 ± 7.36 | 40.20 ± 9.36 | 55.45 ± 21.59 | |
| PH | 113.60 ± 4.04 | 102.80 ± 4.09 | 102.13 ± 19.14 | |
| BN | 13.60 ± 2.60 | 10.40 ± 1.95 | 12.64 ± 3.24 | |
| 2014 | DTB | 90.89 ± 5.37 | 86.56 ± 3.05 | 82.13 ± 13.72 |
| DTF | 111.10 ± 4.33 | 112.00 ± 5.61 | 103.23 ± 14.51 | |
| DTS | 122.67 ± 5.61 | 125.67 ± 2.80 | 116.13 ± 15.50 | |
| LN | 75.00 ± 8.98 | 42.33 ± 4.16 | 59.32 ± 22.90 | |
| PH | 124.88 ± 9.00 | 106.33 ± 4.73 | 111.48 ± 20.38 | |
| BN | 17.25 ± 2.66 | 9.67 ± 2.31 | 15.33 ± 5.13 |
| Trait | Year | DTB | DTF | DTS | LN | PH |
|---|---|---|---|---|---|---|
| DTF | 2013 | 0.969 ** | ||||
| 2014 | 0.983 ** | |||||
| DTS | 2013 | 0.962 ** | 0.989 ** | |||
| 2014 | 0.982 ** | 0.997 ** | ||||
| LN | 2013 | 0.859 ** | 0.903 ** | 0.896 ** | ||
| 2014 | 0.676 ** | 0.685 ** | 0.678 ** | |||
| PH | 2013 | 0.336 ** | 0.35 ** | 0.346 ** | 0.526 ** | |
| 2014 | 0.266 * | 0.268 * | 0.268 * | 0.464 ** | ||
| BN | 2013 | 0.551 ** | 0.634 ** | 0.624 ** | 0.648 ** | 0.219 * |
| 2014 | 0.334 ** | 0.37 ** | 0.372 ** | 0.431 ** | 0.225 * |
| Linkage Group | Total Number of Mapped Markers | Genetic Length (cM) | Physical Length (bp) | Average Interval between Two Markers | |
|---|---|---|---|---|---|
| cM | bp | ||||
| 1 | 212 | 175.9 | 208,403,342 | 0.8 | 983,035 |
| 2 | 229 | 216.1 | 209,216,842 | 0.9 | 913,611 |
| 3 | 115 | 167.3 | 235,685,571 | 1.5 | 2,049,440 |
| 4 | 209 | 277.8 | 359,266,232 | 1.3 | 1,718,977 |
| 5 | 216 | 249.8 | 332,823,101 | 1.2 | 1,540,848 |
| 6 | 77 | 111.6 | 172,193,396 | 1.4 | 2,236,278 |
| 7 | 108 | 155.4 | 178,722,727 | 1.4 | 1,654,840 |
| 8 | 210 | 249.7 | 302,195,534 | 1.2 | 1,439,026 |
| 9 | 127 | 169.9 | 185,552,563 | 1.3 | 1,461,044 |
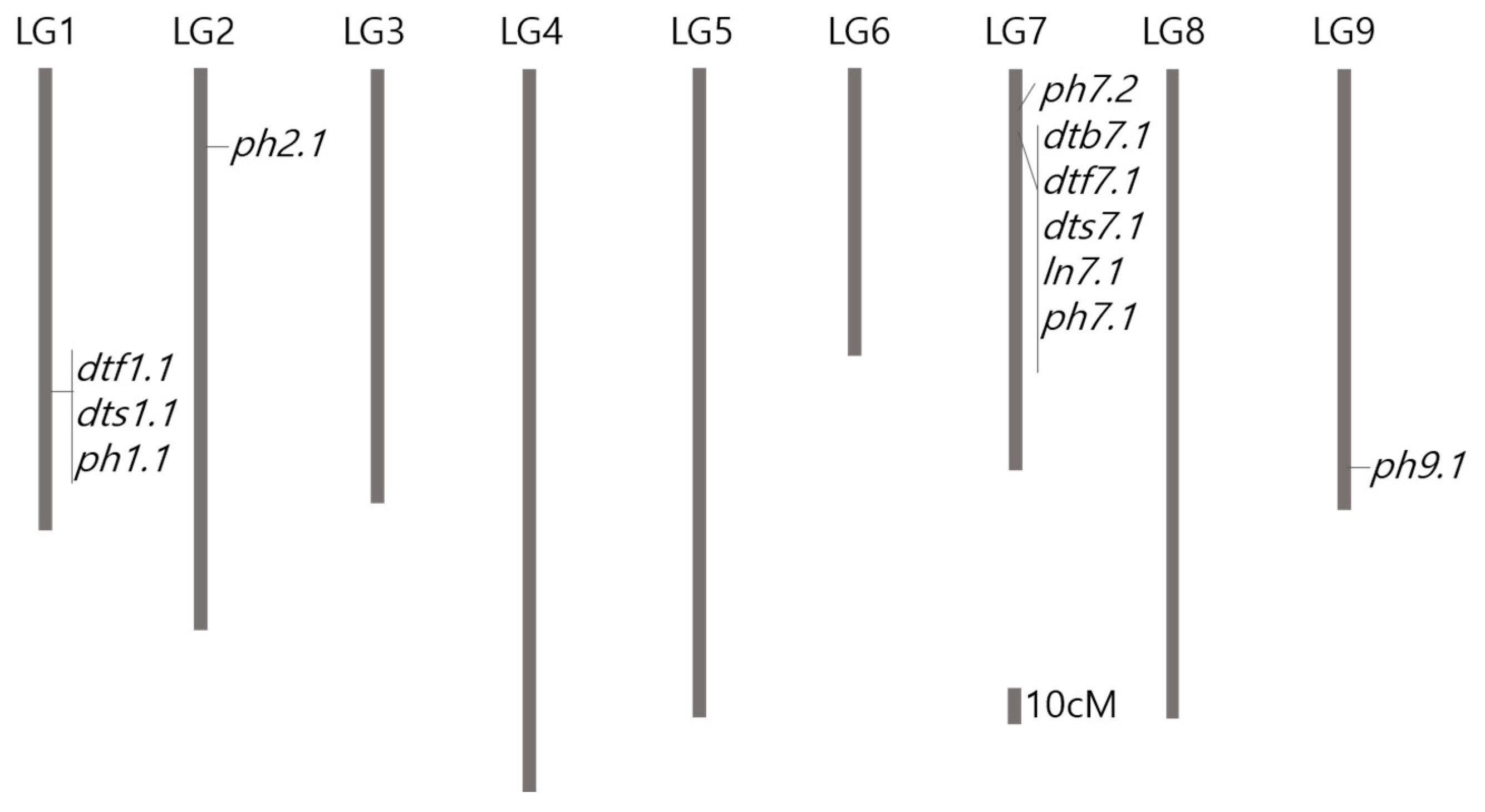
| Trait | QTL | LG | Interval (cM) | Position (cM) a | 2013 | 2014 | Physical Interval (bp) | Physical Position (bp) | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| LOD b | PVE c | Add d | LOD b | PVE c | Add d | |||||||
| DTB | dtb7.1 | 7 | 18.59–29.02 | 20.73 | 17.37 | 15.24 | 10.36 | 16.52 | 11.97 | 9.76 | 159,857,676–166,243,410 | 164,472,862 |
| DTF | dtf1.1 | 1 | 123.11–129.39 | 127.65 | 4.35 | 8.25 | −4.85 | 36,628,878–41,340,346 | 36,628,914 | |||
| dtf7.1 | 7 | 18.59–29.02 | 20.73 | 18.42 | 53.09 | 11.83 | 14.07 | 11.43 | 10.24 | 159,857,676–166,243,410 | 164,472,862 | |
| DTS | dts1.1 | 1 | 123.11–129.39 | 127.65 | 4.36 | 7.63 | −4.78 | 37,964,416–41,340,346 | 36,628,914 | |||
| dts7.1 | 7 | 18.59–29.02 | 20.73 | 19.07 | 55.20 | 12.37 | 14.15 | 11.90 | 11.13 | 159,857,676–166,243,410 | 164,472,862 | |
| LN | ln7.1 | 7 | 18.59–29.02 | 20.73 | 14.00 | 10.82 | 13.28 | 7.67 | 6.14 | 12.18 | 159,857,676–166,243,410 | 164,472,862 |
| PH | ph1.1 | 1 | 110.62–117.50 | 113.29 | 8.18 | 19.42 | −8.49 | 45,487,674–49,165,461 | 50,734,059 | |||
| ph2.1 | 2 | 57.79–68.84 | 66.00 | 4.87 | 11.92 | −6.93 | 169,936,949–179,130,539 | 172,151,268 | ||||
| ph7.1 | 7 | 18.59–29.02 | 20.73 | 14.48 | 37.28 | 11.78 | 159,857,676–166,243,410 | 164,472,930 | ||||
| ph7.2 | 7 | 8.84–16.79 | 14.60 | 4.51 | 15.55 | 7.88 | 166,971,907–175,172,138 | 167,324,134 | ||||
| ph9.1 | 9 | 152.30–167.82 | 164.78 | 5.03 | 13.44 | 7.76 | 10,161,237–30,276,076 | 10,321,730 | ||||
| Gene | ID | Gene Description | Molecular Function | CDS_ID | bp | Ref. a | Alt. b |
|---|---|---|---|---|---|---|---|
| At5g59700 | Y5597_ARATH | Probable receptor-like protein kinase | Protein kinase activity | Lsat_1_v5_gn_7_95041 | 159,881,847 | T | C |
| ZW10 | ZW10_ARATH | Centromere/kinetochore protein zw10 homolog | Cell division | Lsat_1_v5_gn_7_94920 | 159,937,103 | T | C |
| Gene | NIA_CICIN | Nitrate reductase [NADH] | Nitrate assimilation | Lsat_1_v5_gn_7_94901 | 159,961,111 | G | A |
| At4g26790 | GDL66_ARATH | GDSL esterase/lipase | Hydrolase activity | Lsat_1_v5_gn_7_94800 | 160,295,038 | G | T |
| wss2 | YQ77_SCHPO | Ubiquitin and WLM domain-containing metalloprotease | DNA-binding proteins | Lsat_1_v5_gn_7_94721 | 160,394,234 | A | C |
| KAS | KASM_ARATH | 3-oxoacyl-[acyl-carrier-protein] synthase, mitochondrial | 3-oxoacyl-[acyl-carrier-protein] synthase activity | Lsat_1_v5_gn_7_94701 | 160,398,658 | C | T |
| TRZ2 | RNZ2_ARATH | tRNase Z TRZ2, chloroplastic | 3′-tRNA processing endoribonuclease activity | Lsat_1_v5_gn_7_94680 | 160,401,079 | T | C |
| At1g04970 | Y1049_ARATH | Putative BPI/LBP family protein | Lipopolysaccharide binding | Lsat_1_v5_gn_7_94640 | 160,447,537 | T | A |
| GDI1 | GDIR_ARATH | Rho GDP-dissociation inhibitor 1 | Rho GDP-dissociation inhibitor activity | Lsat_1_v5_gn_7_95621 | 162,399,118 | G | T |
| rpoB | RPOB_LACSA | DNA-directed RNA polymerase subunit β | DNA-directed 5′-3′ RNA polymerase activity | Lsat_1_v5_gn_7_95881 | 163,026,039 | T | C |
| ESS2 | ESS2_HUMAN | Splicing factor ESS-2 homolog | Pre-mRNA splicing | Lsat_1_v5_gn_7_96161 | 163,390,527 | C | T |
| Dnajb5 | DNJB5_MOUSE | DnaJ homolog subfamily B member 5 | Chaperone binding | Lsat_1_v5_gn_7_95320 | 163,700,503 | T | C |
| PCMP-H35 | PP373_ARATH | Putative pentatricopeptide repeat-containing protein | Zinc ion binding | Lsat_1_v5_gn_7_96961 | 164,472,930 | G | A |
| PHYC | PHYC_ORYSJ | Phytochrome C | Phosphorelay sensor kinase activity | Lsat_1_v5_gn_7_96941 | 164,640,464 | A | G |
| GNT2 | MGAT2_ARATH | α-1,6-mannosyl-glycoprotein 2-β-N-acetylglucosaminyltransferase | Catalytic activity i | Lsat_1_v5_gn_7_96920 | 164,651,092 | G | A |
| ABC1K7 | AB1K7_ARATH | Protein activity of BC1 complex kinase 7 | Resistance to oxidative stress | Lsat_1_v5_gn_7_96461 | 164,942,842 | G | C |
| To50-2rc | TO50-2rc | Transferase activity | Transferase activity | Lsat_1_v5_gn_7_96441 | 164,951,268 | C | T |
| SHH2 | SHH2_ARATH | Protein SAWADEE homeodomain homolog 2 | Chromatin binding | Lsat_1_v5_gn_7_97960 | 164,983,376 | A | G |
| At3g07680 | P24B2_ARATH | Transmembrane emp24 domain-containing protein p24beta2 | Intracellular protein transport | Lsat_1_v5_gn_7_97980 | 165,012,377 | A | G |
| RLP7 | RLP7_ARATH | Receptor-like protein 7 | Receptor | Lsat_1_v5_gn_7_97661 | 165,751,808 | G | A |
| RLP6 | RLP6_ARATH | Receptor-like protein 6 | Receptor | Lsat_1_v5_gn_7_97581 | 165,785,946 | C | G |
| accD | ACCD_LACSA | Acetyl-coenzyme A carboxylase carboxyl transferase subunit β, chloroplastic | Carboxylase activity | Lsat_1_v5_gn_7_98321 | 165,894,155 | G | T |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, O.N.; Fukushima, K.; Park, H.Y.; Kawabata, S. QTL Analysis of Stem Elongation and Flowering Time in Lettuce Using Genotyping-by-Sequencing. Genes 2021, 12, 947. https://doi.org/10.3390/genes12060947
Lee ON, Fukushima K, Park HY, Kawabata S. QTL Analysis of Stem Elongation and Flowering Time in Lettuce Using Genotyping-by-Sequencing. Genes. 2021; 12(6):947. https://doi.org/10.3390/genes12060947
Chicago/Turabian StyleLee, O New, Keita Fukushima, Han Yong Park, and Saneyuki Kawabata. 2021. "QTL Analysis of Stem Elongation and Flowering Time in Lettuce Using Genotyping-by-Sequencing" Genes 12, no. 6: 947. https://doi.org/10.3390/genes12060947
APA StyleLee, O. N., Fukushima, K., Park, H. Y., & Kawabata, S. (2021). QTL Analysis of Stem Elongation and Flowering Time in Lettuce Using Genotyping-by-Sequencing. Genes, 12(6), 947. https://doi.org/10.3390/genes12060947