
Whole Genome Sequencing Reveals Multiple Linked Genetic Variants on Canine Chromosome 12 Associated with Risk for Symmetrical Lupoid Onychodystrophy (SLO) in the Bearded Collie

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples
2.2. WGS and Variant Calling
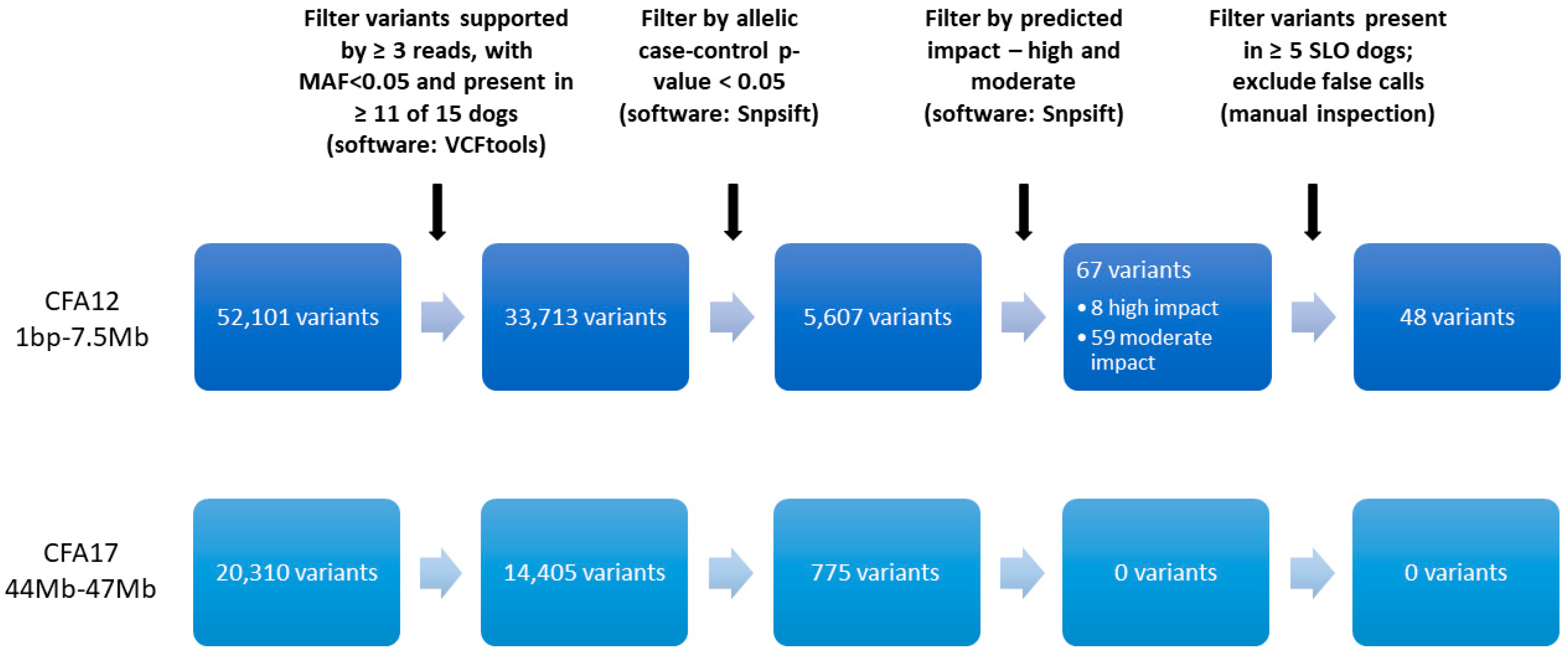
2.3. Variant Filtering for Case-Control Association on 7 SLO and 8 Healthy Controls
2.4. Imputation
2.5. Imputation Accuracy
2.6. TNXB Sequencing
3. Results
3.1. WGS
3.2. Variants from Imputed Dataset
3.3. TNXB Sequencing
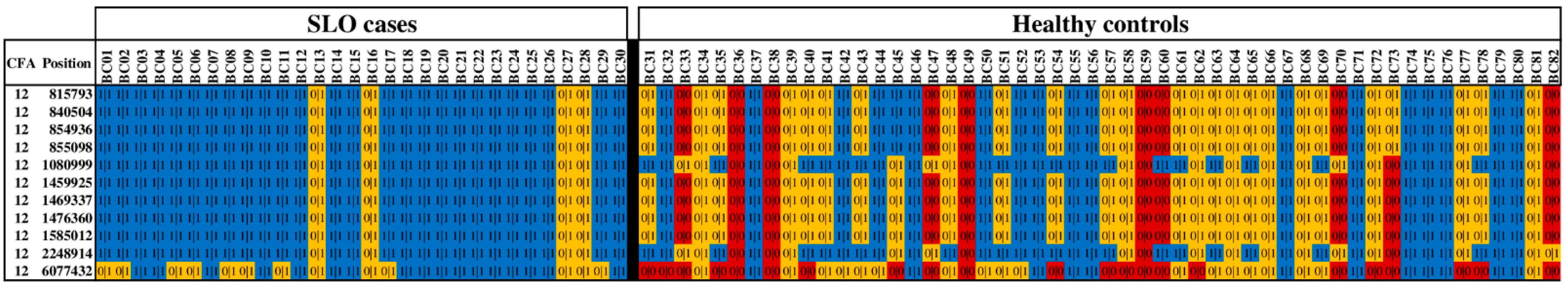
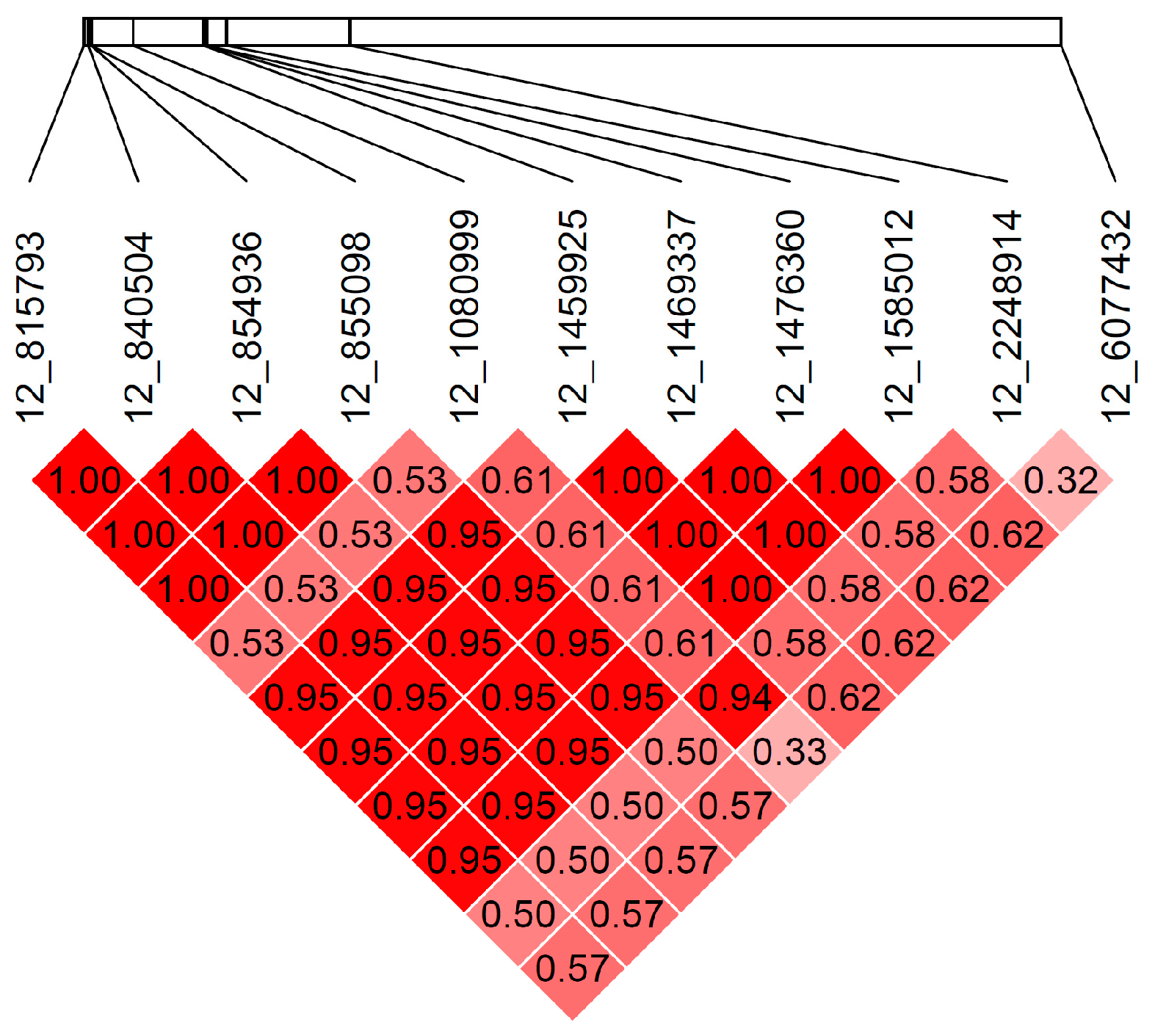
3.4. Additional Variants on CFA12
3.5. CFA17
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ziener, M.L.; Nødtvedt, A. A treatment study of canine symmetrical onychomadesis (symmetrical lupoid onychodystrophy) comparing fish oil and cyclosporine supplementation in addition to a diet rich in omega-3 fatty acids. Acta Vet. Scand. 2014, 56, 66. [Google Scholar] [CrossRef] [Green Version]
- Mueller, R.S.; Rosychuk, R.A.; Jonas, L.D. A retrospective study regarding the treatment of lupoid onychodystrophy in 30 dogs and literature review. J. Am. Anim. Hosp. Assoc. 2003, 39, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Dahlgren, S.; Ziener, M.L.; Lingaas, F. A genome-wide association study identifies a region strongly associated with symmetrical onychomadesis on chromosome 12 in dogs. Anim. Genet. 2016, 47, 708–716. [Google Scholar] [CrossRef] [PubMed]
- Steimer, T.; Bauer, A.; Kienzle, E.; Mueller, R.S. Canine symmetrical lupoid onychomadesis in bearded collies. Vet. Dermatol. 2019, 30, 411–e124. [Google Scholar] [CrossRef] [PubMed]
- Bearded Collie Foundation for Health. BeaCon Open Health Registry Report. Available online: https://www.beaconforhealth.org/Registry_Report_Mar_2019.pdf (accessed on 2 January 2021).
- Mueller, R.S.; Friend, S.; Shipstone, M.A.; Burton, G. Diagnosis of claw disease—A prospective study of 24 dogs. Vet. Dermatol. 2000, 11, 133–141. [Google Scholar] [CrossRef]
- Ziener, M.L.; Bettenay, S.V.; Mueller, R.S. Symmetrical onychomadesis in Norwegian Gordon and English setters. Vet. Dermatol. 2008, 19, 88–94. [Google Scholar] [CrossRef]
- Gershony, L.C.; Belanger, J.M.; Short, A.D.; Le, M.; Hytönen, M.K.; Lohi, H.; Famula, T.R.; Kennedy, L.J.; Oberbauer, A.M. DLA class II risk haplotypes for autoimmune diseases in the bearded collie offer insight to autoimmunity signatures across dog breeds. Canine Genet. Epidemiol. 2019, 6, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilbe, M.; Ziener, M.L.; Aronsson, A.; Harlos, C.; Sundberg, K.; Norberg, E.; Andersson, L.; Lindblad-Toh, K.; Hedhammar, Å.; Andersson, G. DLA class II alleles are associated with risk for canine symmetrical lupoid onychodystropy (SLO). PLoS ONE 2010, 5, e12332. [Google Scholar] [CrossRef]
- Ziener, M.L.; Dahlgren, S.; Thoresen, S.I.; Lingaas, F. Genetics and epidemiology of hypothyroidism and symmetrical onychomadesis in the Gordon setter and the English setter. Canine Genet. Epidemiol. 2015, 2, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gershony, L.C.; Belanger, J.M.; Hytönen, M.K.; Lohi, H.; Oberbauer, A.M. Novel Locus Associated with Symmetrical Lupoid Onychodystrophy in the Bearded Collie. Genes 2019, 10, 635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindblad-Toh, K.; Wade, C.M.; Mikkelsen, T.S.; Karlsson, E.K.; Jaffe, D.B.; Kamal, M.; Clamp, M.; Chang, J.L.; Kulbokas, E.J.; Zody, M.C.; et al. Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature 2005, 438, 803–819. [Google Scholar] [CrossRef]
- Sutter, N.B.; Eberle, M.A.; Parker, H.G.; Pullar, B.J.; Kirkness, E.F.; Kruglyak, L.; Ostrander, E.A. Extensive and breed-specific linkage disequilibrium in Canis familiaris. Genome Res. 2004, 14, 2388–2396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seddon, J.M.; Berggren, K.T.; Fleeman, L.M. Evolutionary history of DLA class II haplotypes in canine diabetes mellitus through single nucleotide polymorphism genotyping. Tissue Antigens 2010, 75, 218–226. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.; Li, D.; Li, C.; Muehleisen, B.; Radek, K.A.; Park, H.J.; Jiang, Z.; Li, Z.; Lei, H.; Quan, Y.; et al. The Antimicrobial Protein REG3A Regulates Keratinocyte Proliferation and Differentiation after Skin Injury. Immunity 2012, 37, 74–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayward, J.J.; White, M.E.; Boyle, M.; Shannon, L.M.; Casal, M.L.; Castelhano, M.G.; Center, S.A.; Meyers-Wallen, V.N.; Simpson, K.W.; Sutter, N.B.; et al. Imputation of canine genotype array data using 365 whole-genome sequences improves power of genome-wide association studies. PLoS Genet. 2019, 15, e1008003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedenberg, S.G.; Meurs, K.M. Genotype imputation in the domestic dog. Mamm. Genome 2016, 27, 485–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenkins, C.A.; Aguirre, G.; André, C.; Bannasch, D.; Becker, D.; Davis, B.; Drögemüller, C.; Ekenstedt, K.; Faller, K.; Forman, O.; et al. Improving the resolution of canine genome-wide association studies using genotype imputation: A study of two breeds. Anim. Genet. 2021. [Google Scholar] [CrossRef] [PubMed]
- Rincon, G.; Tengvall, K.; Belanger, J.M.; Lagoutte, L.; Medrano, J.F.; Andre, C.; Thomas, A.; Lawley, C.T.; Hansen, M.S.; Lindblad-Toh, K.; et al. Comparison of buccal and blood-derived canine DNA, either native or whole genome amplified, for array-based genome-wide association studies. BMC Res. Notes 2011, 4, 226. [Google Scholar] [CrossRef] [PubMed]
- Auxilia, S.T.; Hill, P.B.; Thoday, K.L. Canine symmetrical lupoid onychodystrophy: A retrospective study with particular reference to management. J. Small Anim. Pract. 2001, 42, 82–87. [Google Scholar] [CrossRef]
- O’Connor, B.D.; Van der Auwera, G.A. Genomics in the Cloud: Using Docker, GATK, and WDL in Terra, 1st ed.; O’Reilly Media: Sebastopol, CA, USA, 2020. [Google Scholar]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 8 August 2020).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Howe, K.L.; Achuthan, P.; Allen, J.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Azov, A.G.; Bennett, R.; Bhai, J.; et al. Ensembl 2021. Nucleic Acids Res. 2020, 49, D884–D891. [Google Scholar] [CrossRef]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Subgroup, G.P.D.P. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picard Toolkit. Broad Institute, GitHub Repository. 2019. Available online: http://broadinstitute.github.io/picard/ (accessed on 9 August 2020).
- Cingolani, P.; Platts, A.; Wang le, L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.; Daly, M.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Ruden, D.; Cingolani, P.; Patel, V.; Coon, M.; Nguyen, T.; Land, S.; Lu, X. Using Drosophila melanogaster as a Model for Genotoxic Chemical Mutational Studies with a New Program, SnpSift. Front. Genet. 2012, 3, 35. [Google Scholar] [CrossRef] [Green Version]
- Cingolani, P. SnpEff & SnpSift Documentation. Available online: http://pcingola.github.io/SnpEff/ss_casecontrol/ (accessed on 8 October 2020).
- Ziegler, A.; Van Steen, K.; Wellek, S. Investigating Hardy–Weinberg equilibrium in case–control or cohort studies or meta-analysis. Breast Cancer Res. Treat. 2011, 128, 197–201. [Google Scholar] [CrossRef]
- Browning, S.R.; Browning, B.L. Rapid and Accurate Haplotype Phasing and Missing-Data Inference for Whole-Genome Association Studies By Use of Localized Haplotype Clustering. Am. J. Hum. Genet. 2007, 81, 1084–1097. [Google Scholar] [CrossRef] [Green Version]
- Browning, B.L. Beagle 5.1. Available online: https://faculty.washington.edu/browning/beagle/beagle5.112Aug19.pdf (accessed on 8 November 2020).
- Lewis, T.W.; Abhayaratne, B.M.; Blott, S.C. Trends in genetic diversity for all Kennel Club registered pedigree dog breeds. Canine Genet. Epidemiol. 2015, 2, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, C.L.; Bhérer, C.; Morrow, B.E.; Boyko, A.R.; Auton, A. A Pedigree-Based Map of Recombination in the Domestic Dog Genome. G3 (Bethesda) 2016, 6, 3517–3524. [Google Scholar] [CrossRef] [Green Version]
- Browning, B.L. Conform-gt. Available online: https://faculty.washington.edu/browning/conform-gt.html (accessed on 18 June 2021).
- Browning, B.L.; Zhou, Y.; Browning, S.R. A One-Penny Imputed Genome from Next-Generation Reference Panels. Am. J. Hum. Genet. 2018, 103, 338–348. [Google Scholar] [CrossRef] [Green Version]
- Robinson, J.T.; Thorvaldsdóttir, H.; Wenger, A.M.; Zehir, A.; Mesirov, J.P. Variant Review with the Integrative Genomics Viewer. Cancer Res. 2017, 77, e31–e34. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.; Sims, G.E.; Murphy, S.; Miller, J.R.; Chan, A.P. Predicting the Functional Effect of Amino Acid Substitutions and Indels. PLoS ONE 2012, 7, e46688. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.; Chan, A.P. PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 2015, 31, 2745–2747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, G.M.; Stone, E.A.; Asimenos, G.; Green, E.D.; Batzoglou, S.; Sidow, A. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 2005, 15, 901–913. [Google Scholar] [CrossRef] [Green Version]
- Lowry, R. VassarStats: Website for Statistical Computation. Available online: http://vassarstats.net/odds2x2.html (accessed on 18 June 2021).
- Perdry, H.; Dandine-Roulland, C. Gaston: Genetic Data Handling (QC, GRM, LD, PCA) & Linear Mixed Models. R package version 1.5.7. Available online: https://CRAN.R-project.org/package=gaston (accessed on 29 June 2021).
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2017. [Google Scholar]
- Koressaar, T.; Remm, M. Enhancements and modifications of primer design program Primer3. Bioinformatics 2007, 23, 1289–1291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, M.; Whitmire, S.; Chen, J.; Farrel, A.; Shi, X.; Guo, J.-t. Effects of short indels on protein structure and function in human genomes. Sci. Rep. 2017, 7, 9313. [Google Scholar] [CrossRef] [PubMed]
- Abramowicz, A.; Gos, M. Splicing mutations in human genetic disorders: Examples, detection, and confirmation. J. Appl. Genet. 2018, 59, 253–268. [Google Scholar] [CrossRef] [Green Version]
- Sinclair, R.D.; Wojnarowska, F.; Leigh, I.M.; Dawber, R.P. The basement membrane zone of the nail. Br. J. Dermatol. 1994, 131, 499–505. [Google Scholar] [CrossRef]
- Mao, J.R.; Taylor, G.; Dean, W.B.; Wagner, D.R.; Afzal, V.; Lotz, J.C.; Rubin, E.M.; Bristow, J. Tenascin-X deficiency mimics Ehlers-Danlos syndrome in mice through alteration of collagen deposition. Nat. Genet. 2002, 30, 421–425. [Google Scholar] [CrossRef]
- Kamatani, Y.; Matsuda, K.; Ohishi, T.; Ohtsubo, S.; Yamazaki, K.; Iida, A.; Hosono, N.; Kubo, M.; Yumura, W.; Nitta, K.; et al. Identification of a significant association of a single nucleotide polymorphism in TNXB with systemic lupus erythematosus in a Japanese population. J. Hum. Genet. 2007, 53, 64. [Google Scholar] [CrossRef] [Green Version]
- Bauer, A.; de Lucia, M.; Leuthard, F.; Jagannathan, V.; Leeb, T. Compound heterozygosity for TNXB genetic variants in a mixed-breed dog with Ehlers-Danlos syndrome. Anim. Genet. 2019, 50, 546–549. [Google Scholar] [CrossRef]
- Pénisson-Besnier, I.; Allamand, V.; Beurrier, P.; Martin, L.; Schalkwijk, J.; van Vlijmen-Willems, I.; Gartioux, C.; Malfait, F.; Syx, D.; Macchi, L.; et al. Compound heterozygous mutations of the TNXB gene cause primary myopathy. Neuromuscul. Disord. 2013, 23, 664–669. [Google Scholar] [CrossRef]
- Bexfield, N.H.; Watson, P.J.; Aguirre-Hernandez, J.; Sargan, D.R.; Tiley, L.; Heeney, J.L.; Kennedy, L.J. DLA class II alleles and haplotypes are associated with risk for and protection from chronic hepatitis in the English Springer spaniel. PLoS ONE 2012, 7, e42584. [Google Scholar] [CrossRef]
- Tsai, S.; Santamaria, P. MHC Class II Polymorphisms, Autoreactive T-Cells, and Autoimmunity. Front. Immunol. 2013, 4, 321. [Google Scholar] [CrossRef] [Green Version]
- Holzer, U.; Nepom, G.T. Major Histocompatibility Complex and Autoimmune Disease. In Stem Cell Therapy for Autoimmune Disease; Burt, R.K., Marmont, A.M., Eds.; Landes Bioscience: Georgetown, TX, USA, 2004; pp. 155–162. [Google Scholar]
- Kennedy, L.J.; Barnes, A.; Happ, G.M.; Quinnell, R.J.; Courtenay, O.; Carter, S.D.; Ollier, W.E.R.; Thomson, W. Evidence for extensive DLA polymorphism in different dog populations. Tissue Antigens 2002, 60, 43–52. [Google Scholar] [CrossRef]
- McDevitt, H.; Munson, S.; Ettinger, R.; Wu, A. Multiple roles for tumor necrosis factor-α and lymphotoxin α/β in immunity and autoimmunity. Arthritis Res. Ther. 2002, 4, S141. [Google Scholar] [CrossRef]
- Upadhyay, V.; Fu, Y.-X. Lymphotoxin signalling in immune homeostasis and the control of microorganisms. Nat. Rev. Immunol. 2013, 13, 270–279. [Google Scholar] [CrossRef]
- Agyekum, S.; Church, A.; Sohail, M.; Krausz, T.; Van Noorden, S.; Polak, J.; Cohen, J. Expression of lymphotoxin-beta (LT-β) in chronic inflammatory conditions. J. Pathol. 2003, 199, 115–121. [Google Scholar] [CrossRef]
- Harrison, G.A.; Deane, E.M. cDNA sequence of the lymphotoxin beta chain from a marsupial, Macropus eugenii (Tammar wallaby). J. Interferon Cytokine Res. 1999, 19, 1099–1102. [Google Scholar] [CrossRef]
- Chuan, J.; He, S.; Xie, T.; Wang, G.; Yang, Z. Characterization of guanine nucleotide exchange activity of DH domain of human FGD2. Protein Expr. Purif. 2020, 176, 105693. [Google Scholar] [CrossRef] [PubMed]
- Billard, M.J.; Gall, B.J.; Richards, K.L.; Siderovski, D.P.; Tarrant, T.K. G protein signaling modulator-3: A leukocyte regulator of inflammation in health and disease. Am. J. Clin. Exp. Immunol. 2014, 3, 97–106. [Google Scholar]
- Blumer, J.T.; Han, J.Y.; Yang, G.H.; Lodh, S.; Fontaine, D.A.; Davis, D.B. Tcf19 Knockout Mouse Islets Have Increased Stress-related Gene Expression and Reduced Proliferative Capacity. FASEB J. 2020, 34, 1. [Google Scholar] [CrossRef]
- Cheung, Y.H.; Watkinson, J.; Anastassiou, D. Conditional meta-analysis stratifying on detailed HLA genotypes identifies a novel type 1 diabetes locus around TCF19 in the MHC. Hum. Genet. 2011, 129, 161–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CFA12 Position | Variant Rs Number * | Gene | All Transcripts Affected? | Variant Type | GERP Conservation Score § | SLO (N = 30) | Controls (N = 52) | OR (95% CI) | p-Value ¶ | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| HO ALT | HET | HO ALT | HET | ||||||||
| 815793 | rs851051888 | C12H6orf15 | Yes | Missense | 0.16 | 26 | 4 | 18 | 25 | 12.3 (3.7–40.7) | 0.000007 |
| 840504 | rs851625125 | ENSCAFG00000041540 | Yes | Splice acceptor | −0.15 | 26 | 4 | 18 | 25 | 12.3 (3.7–40.7) | 0.000007 |
| 854936 | Novel | TCF19 | Yes | Frameshift | NA | 26 | 4 | 18 | 25 | 12.3 (3.7–40.7) | 0.000007 |
| 855098 | Novel | TCF19 | Yes | Frameshift | NA | 26 | 4 | 18 | 25 | 12.3 (3.7–40.7) | 0.000007 |
| 1080999 | rs852946032 | LTB | Yes | Missense | 0.16 | 26 | 4 | 32 | 15 | 4.1 (1.2–13.4) | 0.022579 |
| 1459925 | rs22185869 | TNXB | Yes | Missense | −0.86 | 26 | 4 | 17 | 25 | 13.4 (4.0–44.5) | 0.000002 |
| 1469337 | rs8493203 | TNXB | Yes | Missense | −4.06 | 26 | 4 | 17 | 25 | 13.4 (4.0–44.5) | 0.000002 |
| 1476360 | rs853176058 | TNXB | Yes | Missense | 2.66 | 26 | 4 | 17 | 25 | 13.4 (4.0–44.5) | 0.000002 |
| 1585012 | rs851873877 | GPSM3 | No | Frameshift | 0.16 | 26 | 4 | 17 | 25 | 13.4 (4.0–44.5) | 0.000002 |
| 2248914 | rs851008370 | HLA-DQB2 | No | Missense | 0.16 | 26 | 4 | 32 | 15 | 4.1 (1.2–13.4) | 0.022579 |
| 6077432 | rs852291453 | FGD2 | No | Missense | 0.41 | 17 | 13 | 12 | 18 | 4.4 (1.7–11.5) | 0.003677 |
| Genotype at Each Variant Location | SLO (N = 42) | Controls (N = 46) | OR (95% CI) | p-Value ¶ | DLA Class II Risk Haplotypes 1 | ||
|---|---|---|---|---|---|---|---|
| 12:1459925 | 12:1469337 | 12:1476360 | |||||
| GG | GG | TT | 1 | 8 | 0.12 (0.01–0.97) | 0.03139 | No risk haplotypes |
| GA | GC | TC | 5 | 23 | 0.14 (0.05–0.41) | 0.00018 | 1 risk haplotype |
| AA | CC | CC | 36 | 15 | 12.4 (4.29–35.85) | 4.1 × 10−7 | 2 risk haplotypes |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gershony, L.C.; Belanger, J.M.; Hytönen, M.K.; Lohi, H.; Oberbauer, A.M. Whole Genome Sequencing Reveals Multiple Linked Genetic Variants on Canine Chromosome 12 Associated with Risk for Symmetrical Lupoid Onychodystrophy (SLO) in the Bearded Collie. Genes 2021, 12, 1265. https://doi.org/10.3390/genes12081265
Gershony LC, Belanger JM, Hytönen MK, Lohi H, Oberbauer AM. Whole Genome Sequencing Reveals Multiple Linked Genetic Variants on Canine Chromosome 12 Associated with Risk for Symmetrical Lupoid Onychodystrophy (SLO) in the Bearded Collie. Genes. 2021; 12(8):1265. https://doi.org/10.3390/genes12081265
Chicago/Turabian StyleGershony, Liza C., Janelle M. Belanger, Marjo K. Hytönen, Hannes Lohi, and Anita M. Oberbauer. 2021. "Whole Genome Sequencing Reveals Multiple Linked Genetic Variants on Canine Chromosome 12 Associated with Risk for Symmetrical Lupoid Onychodystrophy (SLO) in the Bearded Collie" Genes 12, no. 8: 1265. https://doi.org/10.3390/genes12081265
APA StyleGershony, L. C., Belanger, J. M., Hytönen, M. K., Lohi, H., & Oberbauer, A. M. (2021). Whole Genome Sequencing Reveals Multiple Linked Genetic Variants on Canine Chromosome 12 Associated with Risk for Symmetrical Lupoid Onychodystrophy (SLO) in the Bearded Collie. Genes, 12(8), 1265. https://doi.org/10.3390/genes12081265