
Variants Affecting the C-Terminal Tail of UNC93B1 Are Not a Common Risk Factor for Systemic Lupus Erythematosus

 , , , , ,
, , , , , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient Selection and DNA Extraction
2.2. UNC93B1 Targeted Sanger Sequencing
2.3. Whole Genome Sequencing
2.4. Gene Analysis
3. Results
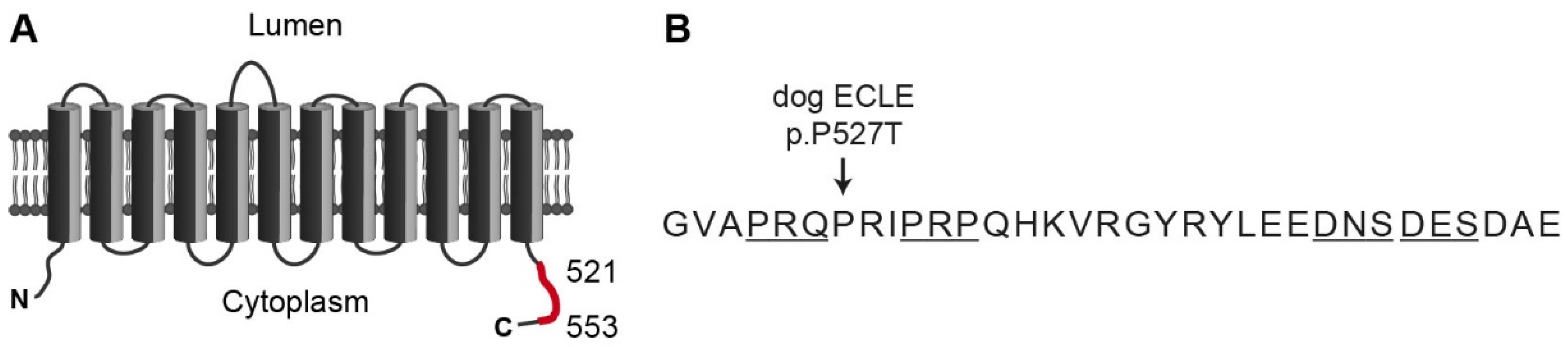
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rahman, A.; Isenberg, D.A. Systemic lupus erythematosus. N. Engl. J. Med. 2008, 358, 929–939. [Google Scholar] [CrossRef] [Green Version]
- Domeier, P.P.; Schell, S.L.; Rahman, Z.S. Spontaneous germinal centers and autoimmunity. Autoimmunity 2017, 50, 4–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitacre, C.C. Sex differences in autoimmune disease. Nat. Immunol. 2001, 2, 777–780. [Google Scholar] [CrossRef] [PubMed]
- Goulielmos, G.N.; Zervou, M.I.; Vazgiourakis, V.M.; Ghodke-Puranik, Y.; Garyfallos, A.; Niewold, T.B. The genetics and molecular pathogenesis of systemic lupus erythematosus (SLE) in populations of different ancestry. Gene 2018, 668, 59–72. [Google Scholar] [CrossRef]
- Fike, A.J.; Elcheva, I.; Rahman, Z.S.M. The Post-GWAS Era: How to Validate the Contribution of Gene Variants in Lupus. Curr. Rheumatol. Rep. 2019, 21, 3. [Google Scholar] [CrossRef] [PubMed]
- Deane, J.A.; Pisitkun, P.; Barrett, R.S.; Feigenbaum, L.; Town, T.; Ward, J.M.; Flavell, R.A.; Bolland, S. Control of toll-like receptor 7 expression is essential to restrict autoimmunity and dendritic cell proliferation. Immunity 2007, 27, 801–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pisitkun, P.; Deane, J.A.; Difilippantonio, M.J.; Tarasenko, T.; Satterthwaite, A.B.; Bolland, S. Autoreactive B cell responses to RNA-related antigens due to TLR7 gene duplication. Science 2006, 312, 1669–1672. [Google Scholar] [CrossRef]
- Subramanian, S.; Tus, K.; Li, Q.Z.; Wang, A.; Tian, X.H.; Zhou, J.; Liang, C.; Bartov, G.; McDaniel, L.D.; Zhou, X.J.; et al. A Tlr7 translocation accelerates systemic autoimmunity in murine lupus. Proc. Natl. Acad. Sci. USA 2006, 103, 9970–9975. [Google Scholar] [CrossRef] [Green Version]
- Souyris, M.; Cenac, C.; Azar, P.; Daviaud, D.; Canivet, A.; Grunenwald, S.; Pienkowski, C.; Chaumeil, J.; Mejía, J.E.; Guéry, J.C. TLR7 escapes X chromosome inactivation in immune cells. Sci. Immunol. 2018, 3, eaap8855. [Google Scholar] [CrossRef] [Green Version]
- Shen, N.; Fu, Q.; Deng, Y.; Qian, X.; Zhao, J.; Kaufman, K.M.; Wu, Y.L.; Yu, C.Y.; Tang, Y.; Chen, J.Y.; et al. Sex-specific association of X-linked Toll-like receptor 7 (TLR7) with male systemic lupus erythematosus. Proc. Natl. Acad. Sci. USA 2010, 107, 15838–15843. [Google Scholar] [CrossRef] [Green Version]
- Raafat, I.I.; El Guindy, N.; Shahin, R.M.H.; Samy, L.A.; El Refai, R.M. Toll-like receptor 7 gene single nucleotide polymorphisms and the risk for systemic lupus erythematosus: A case-control study. Z. Rheumatol. 2018, 77, 416–420. [Google Scholar] [CrossRef]
- Yasutomo, K.; Horiuchi, T.; Kagami, S.; Tsukamoto, H.; Hashimura, C.; Urushihara, M.; Kuroda, Y. Mutation of DNASE1 in people with systemic lupus erythematosus. Nat. Genet. 2001, 28, 313–314. [Google Scholar] [CrossRef] [PubMed]
- Rice, G.; Newman, W.G.; Dean, J.; Patrick, T.; Parmar, R.; Flintoff, K.; Robins, P.; Harvey, S.; Hollis, T.; O’Hara, A.; et al. Heterozygous mutations in TREX1 cause familial chilblain lupus and dominant Aicardi-Goutieres syndrome. Am. J. Hum. Genet. 2007, 80, 811–815. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-M.; Brinkmann, M.M.; Paquet, M.-E.; Ploegh, H.L. UNC93B1 delivers nucleotide-sensing toll-like receptors to endolysosomes. Nature 2008, 452, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Casrouge, A.; Zhang, S.Y.; Eidenschenk, C.; Jouanguy, E.; Puel, A.; Yang, K.; Alcais, A.; Picard, C.; Mahfoufi, N.; Nicolas, N.; et al. Herpes simplex virus encephalitis in human UNC-93B deficiency. Science 2006, 314, 308–312. [Google Scholar] [CrossRef]
- Tabeta, K.; Hoebe, K.; Janssen, E.M.; Du, X.; Georgel, P.; Crozat, K.; Mudd, S.; Mann, N.; Sovath, S.; Goode, J.; et al. The Unc93b1 mutation 3d disrupts exogenous antigen presentation and signaling via Toll-like receptors 3, 7 and 9. Nat. Immunol. 2006, 7, 156–164. [Google Scholar] [CrossRef]
- Ishida, H.; Asami, J.; Zhang, Z.; Nishizawa, T.; Shigematsu, H.; Ohto, U.; Shimizu, T. Cryo-EM structures of Toll-like receptors in complex with UNC93B1. Nat. Struct. Mol. Biol. 2021, 28, 173–180. [Google Scholar] [CrossRef]
- Majer, O.; Liu, B.; Kreuk, L.S.M.; Krogan, N.; Barton, G.M. UNC93B1 recruits syntenin-1 to dampen TLR7 signaling and prevent autoimmunity. Nature 2019, 575, 366–370. [Google Scholar] [CrossRef]
- Saitoh, S.; Miyake, K. Regulatory molecules required for nucleotide-sensing Toll-like receptors. Immunol. Rev. 2009, 227, 32–43. [Google Scholar] [CrossRef]
- Majer, O.; Liu, B.; Woo, B.J.; Kreuk, L.S.M.; Van Dis, E.; Barton, G.M. Release from UNC93B1 reinforces the compartmentalized activation of select TLRs. Nature 2019, 575, 371–374. [Google Scholar] [CrossRef]
- Fukui, R.; Saitoh, S.; Kanno, A.; Onji, M.; Shibata, T.; Ito, A.; Onji, M.; Matsumoto, M.; Akira, S.; Yoshida, N.; et al. Unc93B1 restricts systemic lethal inflammation by orchestrating Toll-like receptor 7 and 9 trafficking. Immunity 2011, 35, 69–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leeb, T.; Leuthard, F.; Jagannathan, V.; Kiener, S.; Letko, A.; Roosje, P.; Welle, M.M.; Gailbreath, K.L.; Cannon, A.; Linek, M.; et al. A missense variant affecting the C-terminal tail of UNC93B1 in dogs with exfoliative cutaneous lupus erythematosus (ECLE). Genes 2020, 11, 159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olivry, T.; Linder, K.E.; Banovic, F. Cutaneous lupus erythematosus in dogs: A comprehensive review. BMC Vet. Res. 2018, 14, 132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vroom, M.W.; Theaker, A.J.; Rest, J.R.; White, S.D. Case report: Lupoid dermatosis in 5 German short-hair pointer. Vet. Dermatol. 1995, 6, 93–98. [Google Scholar] [CrossRef]
- Bryden, S.L.; White, S.D.; Dunston, S.M.; Burrows, A.K.; Olivry, T. Clinical, histopathological and immunological characteristics of exfoliative cutaneous lupus erythematosus in 25 German short-haired pointers. Vet. Dermatol. 2005, 16, 239–252. [Google Scholar] [CrossRef]
- Mauldin, E.A.; Morris, D.O.; Brown, D.C.; Casal, M.L. Exfoliative cutaneous lupus erythematosus in German shorthaired pointer dogs: Disease development, progression and evaluation of three immunomodulatory drugs (ciclosporin, hydroxychloroquine, and adalimumab) in a controlled environment. Vet. Dermatol. 2010, 21, 373–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chizzolini, C.; Cohen, C.D.; Eisenberger, U.; Hauser, T.; Hunziker, T.; Leimgruber, A.; Pechula, M.; Ribi, C.; Stoll, T.; Trendelenburg, M.; et al. Towards the Swiss systemic lupus erythematosus cohort study (SSCS). Rev. Med. Suisse 2009, 5, 808–811. [Google Scholar] [PubMed]
- Ribi, C.; Trendelenburg, M.; Gayet-Ageron, A.; Cohen, C.D.; Dayer, E.; Eisenberger, U.; Hauser, T.; Hunziker, T.; Leimgruber, A.; Lindner, G.; et al. The Swiss Systemic lupus erythematosus Cohort Study (SSCS)-I-Cross-sectional analysis of clinical characteristics and treatments across different medical disciplines in Switzerland. Swiss Med. Wkly. 2014, 144, w13990. [Google Scholar] [CrossRef] [Green Version]
- Meier, A.L.; Bodmer, N.S.; Wirth, C.; Bachmann, L.M.; Ribi, C.; Pröbstel, A.K.; Waeber, D.; Jelcic, I.; Steiner, U.C.; Swiss SLE Cohort Study (SSCS). Neuro-psychiatric manifestations in patients with systemic lupus erythematosus: A systematic review and results from the Swiss lupus cohort study. Lupus 2021, 21, 9612033211025636. [Google Scholar]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997v2. [Google Scholar]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrison, E.; Marth, G. Haplotype-based variant detection from short-read sequencing. arXiv 2012, arXiv:1207.3907. [Google Scholar]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
| dbSNP | HGVS-c | HGVS-p | Alternative Allele Count (Frequency) | gnomAD Allele Frequency |
|---|---|---|---|---|
| rs7149 | c.1557C>G | p.Arg519= | 253 (23.6%) | 16.0% |
| rs576491436 | c.1629G>A | p.Glu543= | 1 (0.1%) | 5 × 10−4 |
| rs1308430306 | c.1651G>A | p.Asp551Asn | 1 (0.1%) | 8 × 10−6 |
| n.a. | c.1724_1725delinsAG | p.Pro575Gln | 8 (0.7%) | n.a. |
| rs2375182 | c.1768G>T | p.Gly590Trp | 4 (0.4%) | 0.4% |
| rs964738111 | c.1777G>A | p.Gly593Arg | 1 (0.1%) | 2 × 10−4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kiener, S.; Ribi, C.; Keller, I.; Chizzolini, C.; Trendelenburg, M.; Huynh-Do, U.; von Kempis, J.; on behalf of Swiss SLE Cohort Study (SSCS); Leeb, T. Variants Affecting the C-Terminal Tail of UNC93B1 Are Not a Common Risk Factor for Systemic Lupus Erythematosus. Genes 2021, 12, 1268. https://doi.org/10.3390/genes12081268
Kiener S, Ribi C, Keller I, Chizzolini C, Trendelenburg M, Huynh-Do U, von Kempis J, on behalf of Swiss SLE Cohort Study (SSCS), Leeb T. Variants Affecting the C-Terminal Tail of UNC93B1 Are Not a Common Risk Factor for Systemic Lupus Erythematosus. Genes. 2021; 12(8):1268. https://doi.org/10.3390/genes12081268
Chicago/Turabian StyleKiener, Sarah, Camillo Ribi, Irene Keller, Carlo Chizzolini, Marten Trendelenburg, Uyen Huynh-Do, Johannes von Kempis, on behalf of Swiss SLE Cohort Study (SSCS), and Tosso Leeb. 2021. "Variants Affecting the C-Terminal Tail of UNC93B1 Are Not a Common Risk Factor for Systemic Lupus Erythematosus" Genes 12, no. 8: 1268. https://doi.org/10.3390/genes12081268
APA StyleKiener, S., Ribi, C., Keller, I., Chizzolini, C., Trendelenburg, M., Huynh-Do, U., von Kempis, J., on behalf of Swiss SLE Cohort Study (SSCS), & Leeb, T. (2021). Variants Affecting the C-Terminal Tail of UNC93B1 Are Not a Common Risk Factor for Systemic Lupus Erythematosus. Genes, 12(8), 1268. https://doi.org/10.3390/genes12081268