
High-Throughput Sequencing to Identify Mutations Associated with Retinal Dystrophies


Abstract
:1. Introduction
2. Material and Methods
2.1. Patients and Family Members
2.2. Ophthalmological Investigations
2.3. DNA Extraction
2.4. Whole Exome Sequencing (WES)
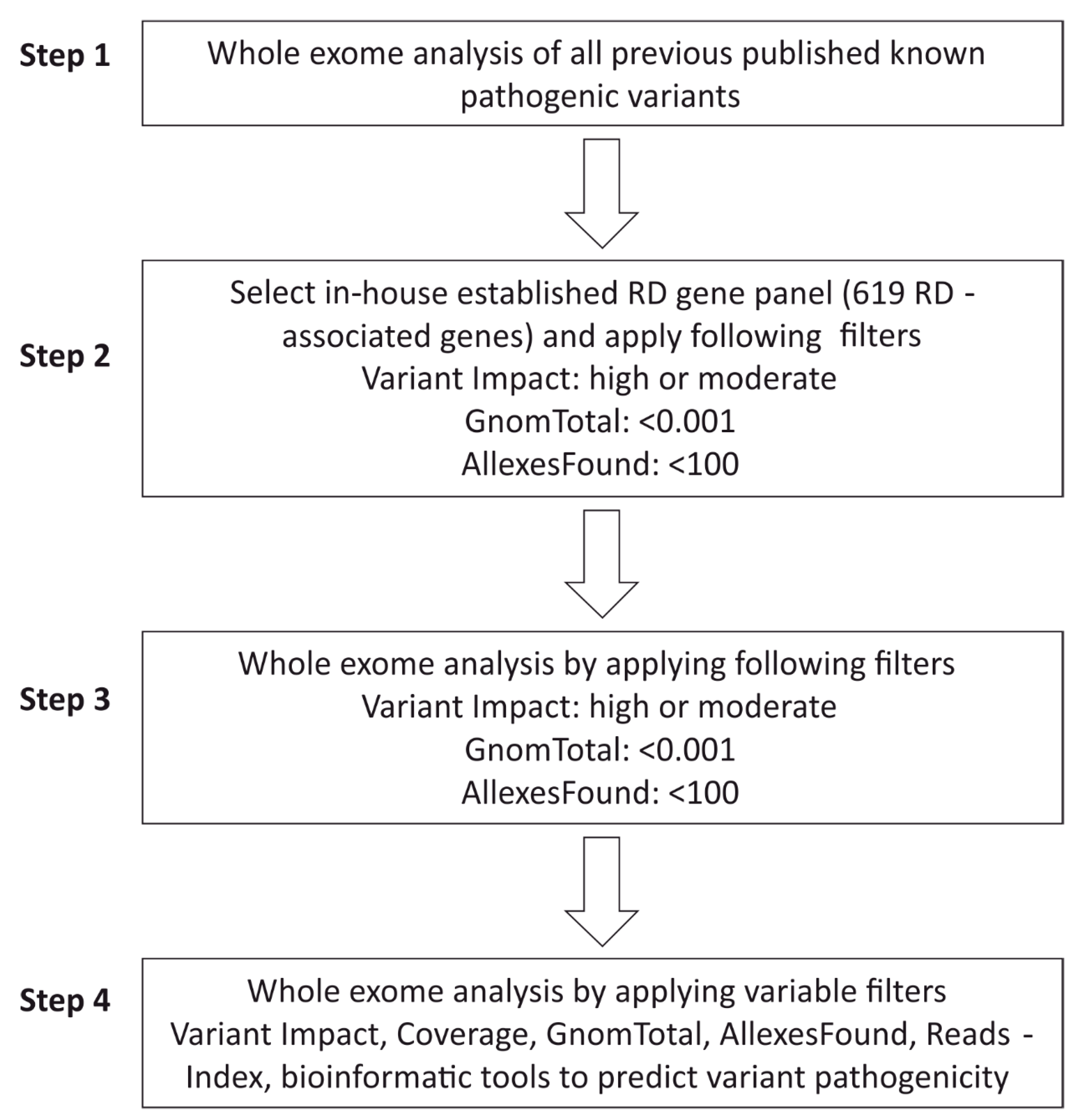
2.5. WES Analysis Pipeline
2.6. Classification of Sequence Variants
- Pathogenic variant: A strong evidence of pathogenicity of the variant was found. This included: (i) The variant was described in literature as a clearly disease-causing mutation. (ii) Novel mutation causes damaging effects on the RNA and protein level, such as canonical splice mutation, nonsense mutation, frameshift mutation, insertion, and deletion. (iii) Mutation was validated by functional studies and segregated within the family.
- Likely pathogenic variant: there is evidence of pathogenicity which included: (i) splice variants were verified by in silico-predicted splice defects. (ii) The novel variant showed low allele frequencies or was predicted as pathogenic using in silico programs (e.g., SIFT, MutationTaster, PolyPhen-2, MutationAssessor). (iii) The variant was confirmed by functional studies or segregation analysis.
- Variant of unsure significance (VUS): There is limited evidence of pathogenicity. This variant does not fulfill the criteria of either pathogenic or benign, or the evidence is conflicting.
- Likely benign variant: There is evidence against pathogenicity, e.g., allele frequency is much higher than expected for the disease or in silico prediction showed conflicting results.
- Benign variant: There is strong evidence against pathogenicity. This variant is probably not a disease-causing mutation, because: (i) The allele frequency is higher than expected (e.g., >1% in GnomAD) for rare genetic diseases. (ii) The variant was observed in healthy populations with inheritance patterns comparable to the affected patient. (iii) Functional studies suggested no damaging effects on RNA or protein level.
2.7. Sanger Sequencing
3. Results
3.1. Patients and Clinical Characterizations
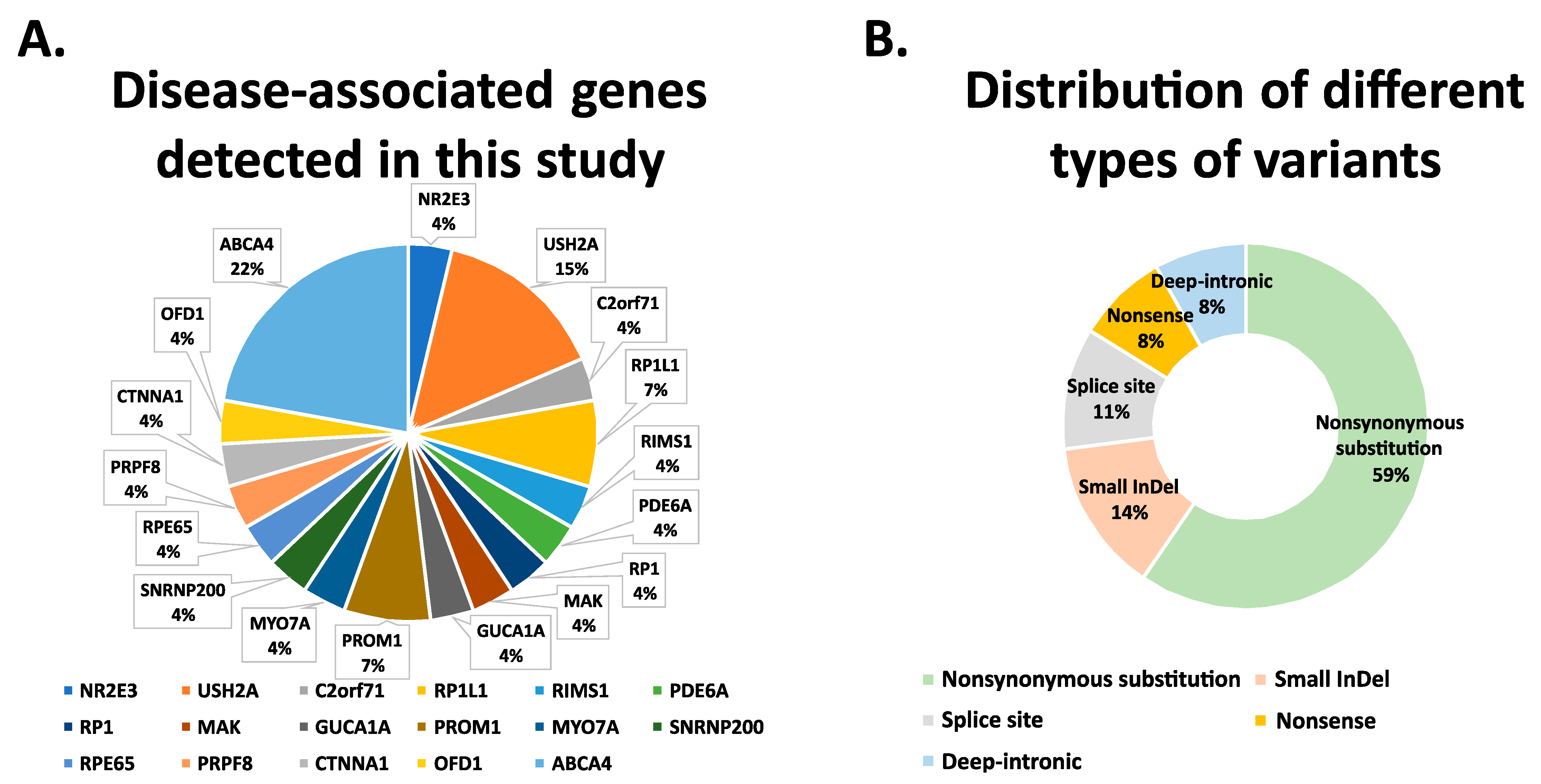
3.2. Whole Exome Sequencing Analysis
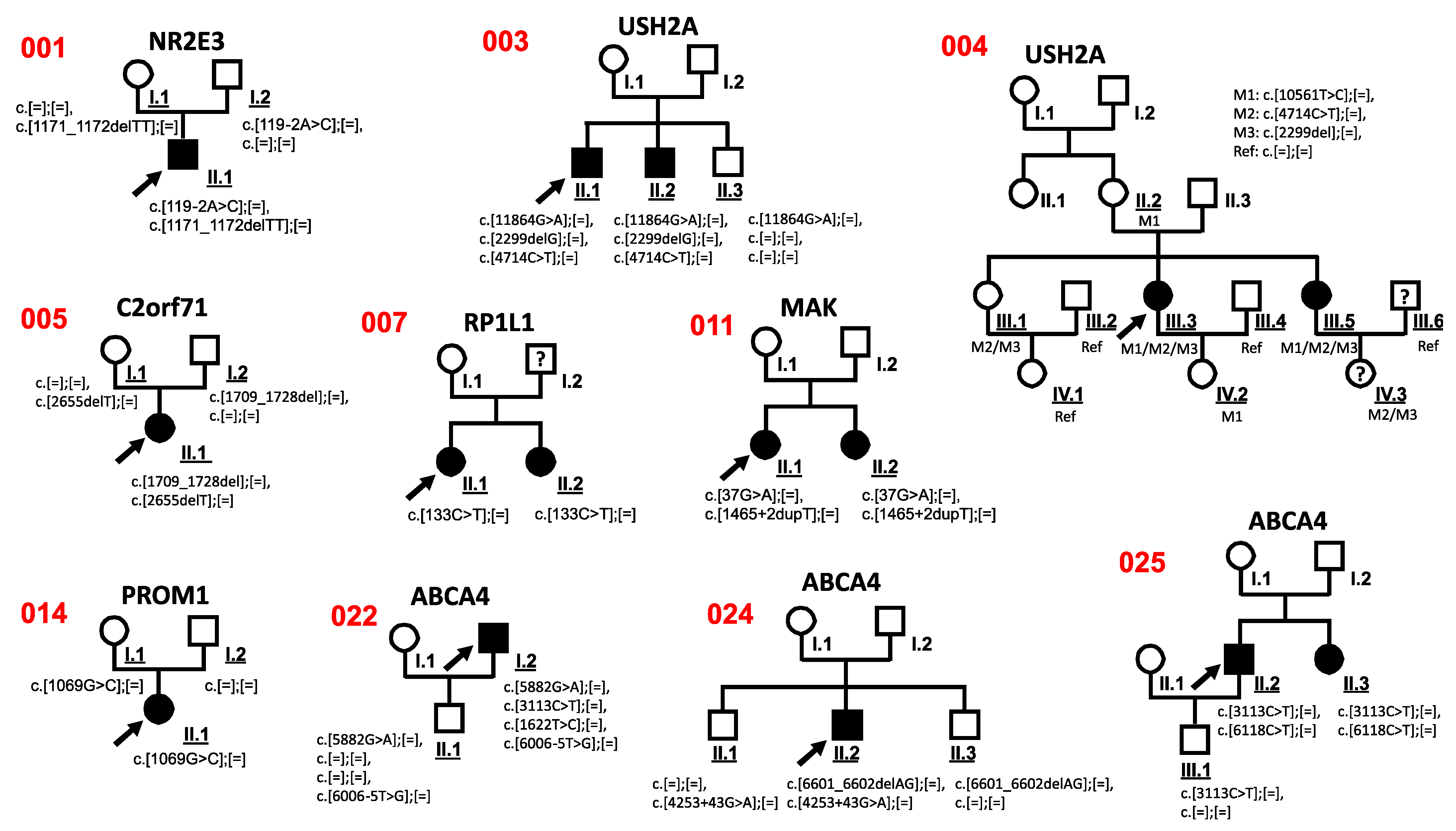
3.3. Co-Segregation Analysis
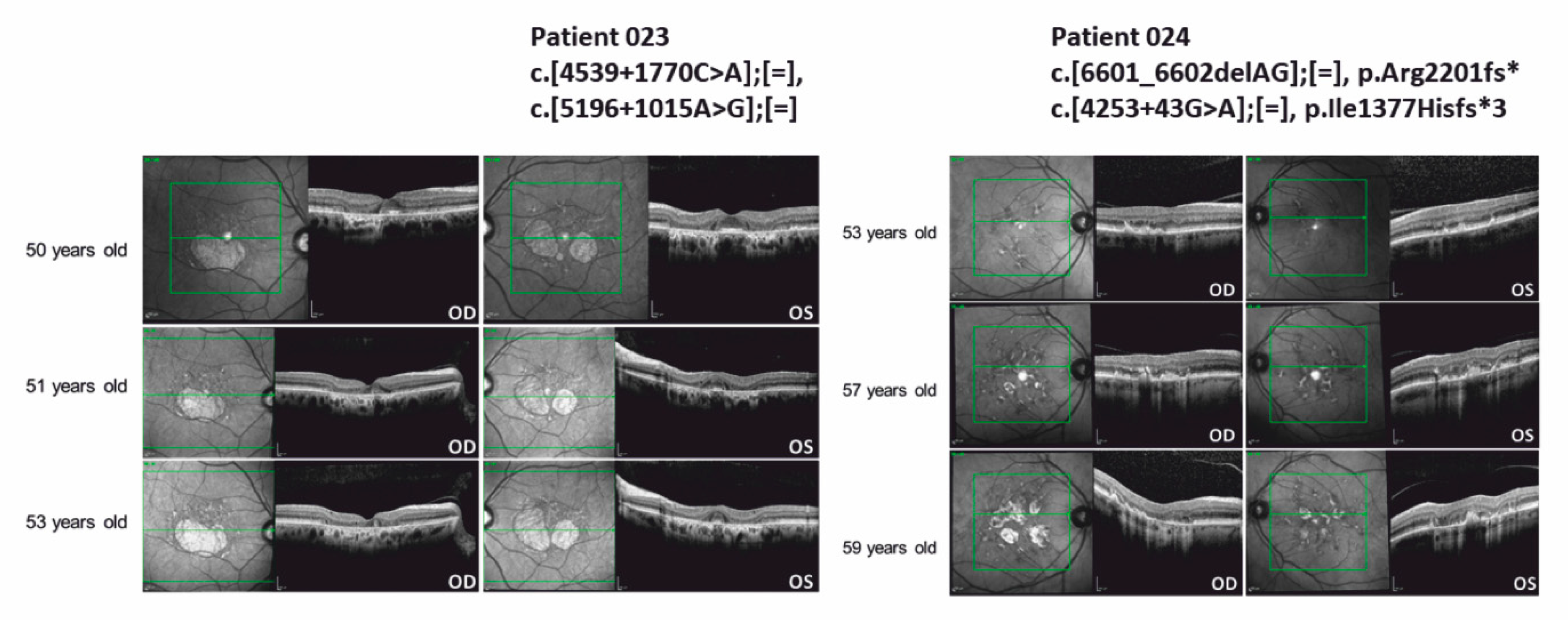
3.4. ABCA4 Deep-Intronic Variants Found in STDG-Patients
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Berger, W.; Kloeckener-Gruissem, B.; Neidhardt, J. The molecular basis of human retinal and vitreoretinal diseases. Prog. Retin. Eye Res. 2010, 29, 335–375. [Google Scholar] [CrossRef] [Green Version]
- Sohocki, M.M.; Daiger, S.P.; Bowne, S.J.; Rodriquez, J.A.; Northrup, H.; Heckenlively, J.R.; Birch, D.G.; Mintz-Hittner, H.; Ruiz, R.S.; Lewis, R.A.; et al. Prevalence of mutations causing retinitis pigmentosa and other inherited retinopathies. Hum. Mutat. 2001, 17, 42–51. [Google Scholar] [CrossRef] [Green Version]
- Bhatia, S.; Kaur, N.; Singh, I.R.; Vanita, V. A novel mutation in MERTK for rod-cone dystrophy in a North Indian family. Can. J. Ophthalmol. 2019, 54, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, S.; Goyal, S.; Singh, I.R.; Singh, D.; Vanita, V. A novel mutation in the PRPF31 in a North Indian adRP family with incomplete penetrance. Doc. Ophthalmol. 2018, 137, 103–119. [Google Scholar] [CrossRef]
- Bainbridge, J.W.; Smith, A.J.; Barker, S.S.; Robbie, S.; Henderson, R.; Balaggan, K.; Viswanathan, A.; Holder, G.E.; Stockman, A.; Tyler, N.; et al. Effect of gene therapy on visual function in Leber’s congenital amaurosis. N. Engl. J. Med. 2008, 358, 2231–2239. [Google Scholar] [CrossRef] [PubMed]
- Maguire, A.M.; Simonelli, F.; Pierce, E.A.; Pugh, E.N., Jr.; Mingozzi, F.; Bennicelli, J.; Banfi, S.; Marshall, K.A.; Testa, F.; Surace, E.M.; et al. Safety and efficacy of gene transfer for Leber’s congenital amaurosis. N. Engl. J. Med. 2008, 358, 2240–2248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pennesi, M.E.; Weleber, R.G.; Yang, P.; Whitebirch, C.; Thean, B.; Flotte, T.R.; Humphries, M.; Chegarnov, E.; Beasley, K.N.; Stout, J.T.; et al. Results at 5 Years After Gene Therapy for RPE65-Deficient Retinal Dystrophy. Hum. Gene Ther. 2018, 29, 1428–1437. [Google Scholar] [CrossRef] [PubMed]
- Prado, D.A.; Acosta-Acero, M.; Maldonado, R.S. Gene therapy beyond luxturna: A new horizon of the treatment for inherited retinal disease. Curr. Opin. Ophthalmol. 2020, 31, 147–154. [Google Scholar] [CrossRef]
- Maguire, A.M.; Russell, S.; Wellman, J.A.; Chung, D.C.; Yu, Z.F.; Tillman, A.; Wittes, J.; Pappas, J.; Elci, O.; Marshall, K.A.; et al. Efficacy, Safety, and Durability of Voretigene Neparvovec-rzyl in RPE65 Mutation-Associated Inherited Retinal Dystrophy: Results of Phase 1 and 3 Trials. Ophthalmology 2019, 126, 1273–1285. [Google Scholar] [CrossRef] [Green Version]
- Russell, S.; Bennett, J.; Wellman, J.A.; Chung, D.C.; Yu, Z.F.; Tillman, A.; Wittes, J.; Pappas, J.; Elci, O.; McCague, S.; et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: A randomised, controlled, open-label, phase 3 trial. Lancet 2017, 390, 849–860. [Google Scholar] [CrossRef]
- Costa, K.A.; Salles, M.V.; Whitebirch, C.; Chiang, J.; Sallum, J.M.F. Gene panel sequencing in Brazilian patients with retinitis pigmentosa. Int. J. Retina Vitreous 2017, 3, 33. [Google Scholar] [CrossRef] [Green Version]
- Glockle, N.; Kohl, S.; Mohr, J.; Scheurenbrand, T.; Sprecher, A.; Weisschuh, N.; Bernd, A.; Rudolph, G.; Schubach, M.; Poloschek, C.; et al. Panel-based next generation sequencing as a reliable and efficient technique to detect mutations in unselected patients with retinal dystrophies. Eur. J. Hum. Genet. 2014, 22, 99–104. [Google Scholar] [CrossRef]
- Tiwari, A.; Bahr, A.; Bahr, L.; Fleischhauer, J.; Zinkernagel, M.S.; Winkler, N.; Barthelmes, D.; Berger, L.; Gerth-Kahlert, C.; Neidhardt, J.; et al. Next generation sequencing based identification of disease-associated mutations in Swiss patients with retinal dystrophies. Sci. Rep. 2016, 6, 28755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almoguera, B.; Li, J.; Fernandez-San Jose, P.; Liu, Y.; March, M.; Pellegrino, R.; Golhar, R.; Corton, M.; Blanco-Kelly, F.; Lopez-Molina, M.I.; et al. Application of Whole Exome Sequencing in Six Families with an Initial Diagnosis of Autosomal Dominant Retinitis Pigmentosa: Lessons Learned. PLoS ONE 2015, 10, e0133624. [Google Scholar] [CrossRef]
- Dan, H.; Huang, X.; Xing, Y.; Shen, Y. Application of targeted panel sequencing and whole exome sequencing for 76 Chinese families with retinitis pigmentosa. Mol. Genet. Genom. Med. 2020, 8, e1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Zhang, J.; Chen, N.; Wang, L.; Zhang, F.; Ma, Z.; Li, G.; Yang, L. Application of Whole Exome and Targeted Panel Sequencing in the Clinical Molecular Diagnosis of 319 Chinese Families with Inherited Retinal Dystrophy and Comparison Study. Genes 2018, 9, 360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albert, S.; Garanto, A.; Sangermano, R.; Khan, M.; Bax, N.M.; Hoyng, C.B.; Zernant, J.; Lee, W.; Allikmets, R.; Collin, R.W.J.; et al. Identification and Rescue of Splice Defects Caused by Two Neighboring Deep-Intronic ABCA4 Mutations Underlying Stargardt Disease. Am. J. Hum. Genet. 2018, 102, 517–527. [Google Scholar] [CrossRef] [Green Version]
- Sangermano, R.; Garanto, A.; Khan, M.; Runhart, E.H.; Bauwens, M.; Bax, N.M.; van den Born, L.I.; Khan, M.I.; Cornelis, S.S.; Verheij, J.; et al. Deep-intronic ABCA4 variants explain missing heritability in Stargardt disease and allow correction of splice defects by antisense oligonucleotides. Genet. Med. 2019, 21, 1751–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulz, H.L.; Grassmann, F.; Kellner, U.; Spital, G.; Ruther, K.; Jagle, H.; Hufendiek, K.; Rating, P.; Huchzermeyer, C.; Baier, M.J.; et al. Mutation Spectrum of the ABCA4 Gene in 335 Stargardt Disease Patients From a Multicenter German Cohort-Impact of Selected Deep Intronic Variants and Common SNPs. Investig. Ophthalmol. Vis. Sci. 2017, 58, 394–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zernant, J.; Lee, W.; Nagasaki, T.; Collison, F.T.; Fishman, G.A.; Bertelsen, M.; Rosenberg, T.; Gouras, P.; Tsang, S.H.; Allikmets, R. Extremely hypomorphic and severe deep intronic variants in the ABCA4 locus result in varying Stargardt disease phenotypes. Cold Spring Harb. Mol. Case Stud. 2018, 4, a002733. [Google Scholar] [CrossRef] [Green Version]
- Owczarek-Lipska, M.; Song, F.; Jaksic, V.; Neidhardt, J. Compound heterozygous RPE65 mutations associated with an early onset autosomal recessive retinitis pigmentosa. J. Gene Med. 2020, 22, e3211. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Haider, N.B.; Jacobson, S.G.; Cideciyan, A.V.; Swiderski, R.; Streb, L.M.; Searby, C.; Beck, G.; Hockey, R.; Hanna, D.B.; Gorman, S.; et al. Mutation of a nuclear receptor gene, NR2E3, causes enhanced S cone syndrome, a disorder of retinal cell fate. Nat. Genet. 2000, 24, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Smaoui, N.; Ayyagari, R.; Stiles, D.; Benhamed, S.; MacDonald, I.M.; Daiger, S.P.; Tumminia, S.J.; Hejtmancik, F.; Wang, X. High-throughput retina-array for screening 93 genes involved in inherited retinal dystrophy. Investig. Ophthalmol. Vis. Sci. 2011, 52, 9053–9060. [Google Scholar] [CrossRef]
- Aller, E.; Larrieu, L.; Jaijo, T.; Baux, D.; Espinos, C.; Gonzalez-Candelas, F.; Najera, C.; Palau, F.; Claustres, M.; Roux, A.F.; et al. The USH2A c.2299delG mutation: Dating its common origin in a Southern European population. Eur. J. Hum. Genet. 2010, 18, 788–793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Wijk, E.; Pennings, R.J.; te Brinke, H.; Claassen, A.; Yntema, H.G.; Hoefsloot, L.H.; Cremers, F.P.; Cremers, C.W.; Kremer, H. Identification of 51 novel exons of the Usher syndrome type 2A (USH2A) gene that encode multiple conserved functional domains and that are mutated in patients with Usher syndrome type II. Am. J. Hum. Genet. 2004, 74, 738–744. [Google Scholar] [CrossRef] [Green Version]
- Dreyer, B.; Brox, V.; Tranebjaerg, L.; Rosenberg, T.; Sadeghi, A.M.; Moller, C.; Nilssen, O. Spectrum of USH2A mutations in Scandinavian patients with Usher syndrome type II. Hum. Mutat. 2008, 29, 451. [Google Scholar] [CrossRef] [PubMed]
- Fujinami, K.; Kameya, S.; Kikuchi, S.; Ueno, S.; Kondo, M.; Hayashi, T.; Shinoda, K.; Machida, S.; Kuniyoshi, K.; Kawamura, Y.; et al. Novel RP1L1 Variants and Genotype-Photoreceptor Microstructural Phenotype Associations in Cohort of Japanese Patients With Occult Macular Dystrophy. Investig. Ophthalmol. Vis. Sci. 2016, 57, 4837–4846. [Google Scholar] [CrossRef] [Green Version]
- Akahori, M.; Tsunoda, K.; Miyake, Y.; Fukuda, Y.; Ishiura, H.; Tsuji, S.; Usui, T.; Hatase, T.; Nakamura, M.; Ohde, H.; et al. Dominant mutations in RP1L1 are responsible for occult macular dystrophy. Am. J. Hum. Genet. 2010, 87, 424–429. [Google Scholar] [CrossRef] [Green Version]
- Ozgul, R.K.; Siemiatkowska, A.M.; Yucel, D.; Myers, C.A.; Collin, R.W.; Zonneveld, M.N.; Beryozkin, A.; Banin, E.; Hoyng, C.B.; van den Born, L.I.; et al. Exome sequencing and cis-regulatory mapping identify mutations in MAK, a gene encoding a regulator of ciliary length, as a cause of retinitis pigmentosa. Am. J. Hum. Genet. 2011, 89, 253–264. [Google Scholar] [CrossRef] [Green Version]
- Sokal, I.; Dupps, W.J.; Grassi, M.A.; Brown, J., Jr.; Affatigato, L.M.; Roychowdhury, N.; Yang, L.; Filipek, S.; Palczewski, K.; Stone, E.M.; et al. A novel GCAP1 missense mutation (L151F) in a large family with autosomal dominant cone-rod dystrophy (adCORD). Investig. Ophthalmol. Vis. Sci. 2005, 46, 1124–1132. [Google Scholar] [CrossRef]
- Yang, Z.; Chen, Y.; Lillo, C.; Chien, J.; Yu, Z.; Michaelides, M.; Klein, M.; Howes, K.A.; Li, Y.; Kaminoh, Y.; et al. Mutant prominin 1 found in patients with macular degeneration disrupts photoreceptor disk morphogenesis in mice. J. Clin. Investig. 2008, 118, 2908–2916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bharadwaj, A.K.; Kasztejna, J.P.; Huq, S.; Berson, E.L.; Dryja, T.P. Evaluation of the myosin VIIA gene and visual function in patients with Usher syndrome type I. Exp. Eye Res. 2000, 71, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Bellur, D.L.; Lu, S.; Zhao, F.; Grassi, M.A.; Bowne, S.J.; Sullivan, L.S.; Daiger, S.P.; Chen, L.J.; Pang, C.P.; et al. Autosomal-dominant retinitis pigmentosa caused by a mutation in SNRNP200, a gene required for unwinding of U4/U6 snRNAs. Am. J. Hum. Genet. 2009, 85, 617–627. [Google Scholar] [CrossRef] [Green Version]
- Gerber, S.; Rozet, J.M.; van de Pol, T.J.; Hoyng, C.B.; Munnich, A.; Blankenagel, A.; Kaplan, J.; Cremers, F.P. Complete exon-intron structure of the retina-specific ATP binding transporter gene (ABCR) allows the identification of novel mutations underlying Stargardt disease. Genomics 1998, 48, 139–142. [Google Scholar] [CrossRef]
- Maugeri, A.; Klevering, B.J.; Rohrschneider, K.; Blankenagel, A.; Brunner, H.G.; Deutman, A.F.; Hoyng, C.B.; Cremers, F.P. Mutations in the ABCA4 (ABCR) gene are the major cause of autosomal recessive cone-rod dystrophy. Am. J. Hum. Genet. 2000, 67, 960–966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allikmets, R.; Shroyer, N.F.; Singh, N.; Seddon, J.M.; Lewis, R.A.; Bernstein, P.S.; Peiffer, A.; Zabriskie, N.A.; Li, Y.; Hutchinson, A.; et al. Mutation of the Stargardt disease gene (ABCR) in age-related macular degeneration. Science 1997, 277, 1805–1807. [Google Scholar] [CrossRef] [Green Version]
- Baux, D.; Larrieu, L.; Blanchet, C.; Hamel, C.; Ben Salah, S.; Vielle, A.; Gilbert-Dussardier, B.; Holder, M.; Calvas, P.; Philip, N.; et al. Molecular and in silico analyses of the full-length isoform of usherin identify new pathogenic alleles in Usher type II patients. Hum. Mutat. 2007, 28, 781–789. [Google Scholar] [CrossRef]
- Sciezynska, A.; Ozieblo, D.; Ambroziak, A.M.; Korwin, M.; Szulborski, K.; Krawczynski, M.; Stawinski, P.; Szaflik, J.; Szaflik, J.P.; Ploski, R.; et al. Next-generation sequencing of ABCA4: High frequency of complex alleles and novel mutations in patients with retinal dystrophies from Central Europe. Exp. Eye Res. 2016, 145, 93–99. [Google Scholar] [CrossRef]
- Nassisi, M.; Mohand-Said, S.; Andrieu, C.; Antonio, A.; Condroyer, C.; Mejecase, C.; Varin, J.; Wohlschlegel, J.; Dhaenens, C.M.; Sahel, J.A.; et al. Prevalence of ABCA4 Deep-Intronic Variants and Related Phenotype in An Unsolved “One-Hit” Cohort with Stargardt Disease. Int. J. Mol. Sci. 2019, 20, 5053. [Google Scholar] [CrossRef] [Green Version]
- van Dijk, E.L.; Auger, H.; Jaszczyszyn, Y.; Thermes, C. Ten years of next-generation sequencing technology. Trends Genet. 2014, 30, 418–426. [Google Scholar] [CrossRef]
- Barbitoff, Y.A.; Polev, D.E.; Glotov, A.S.; Serebryakova, E.A.; Shcherbakova, I.V.; Kiselev, A.M.; Kostareva, A.A.; Glotov, O.S.; Predeus, A.V. Systematic dissection of biases in whole-exome and whole-genome sequencing reveals major determinants of coding sequence coverage. Sci. Rep. 2020, 10, 2057. [Google Scholar] [CrossRef] [Green Version]
- Schwarze, K.; Buchanan, J.; Taylor, J.C.; Wordsworth, S. Are whole-exome and whole-genome sequencing approaches cost-effective? A systematic review of the literature. Genet. Med. 2018, 20, 1122–1130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, Y.S.; Huang, H.D.; Yeh, K.T.; Chang, J.G. Evaluation of whole exome sequencing by targeted gene sequencing and Sanger sequencing. Clin. Chim. Acta 2017, 471, 222–232. [Google Scholar] [CrossRef]
- Bryant, L.; Lozynska, O.; Maguire, A.M.; Aleman, T.S.; Bennett, J. Prescreening whole exome sequencing results from patients with retinal degeneration for variants in genes associated with retinal degeneration. Clin. Ophthalmol. 2018, 12, 49–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carss, K.J.; Arno, G.; Erwood, M.; Stephens, J.; Sanchis-Juan, A.; Hull, S.; Megy, K.; Grozeva, D.; Dewhurst, E.; Malka, S.; et al. Comprehensive Rare Variant Analysis via Whole-Genome Sequencing to Determine the Molecular Pathology of Inherited Retinal Disease. Am. J. Hum. Genet. 2017, 100, 75–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burdick, K.J.; Cogan, J.D.; Rives, L.C.; Robertson, A.K.; Koziura, M.E.; Brokamp, E.; Duncan, L.; Hannig, V.; Pfotenhauer, J.; Vanzo, R.; et al. Limitations of exome sequencing in detecting rare and undiagnosed diseases. Am. J. Med. Genet. A 2020, 182, 1400–1406. [Google Scholar] [CrossRef] [PubMed]
- Yi, Z.; Ouyang, J.; Sun, W.; Li, S.; Xiao, X.; Zhang, Q. Comparative exome sequencing reveals novel candidate genes for retinitis pigmentosa. EBioMedicine 2020, 56, 102792. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lab ID | Gender | Ancestry | Clinical Diagnosis | Disease Causing Gene | Nucleotide Change | Amino Acid Change | GnomAD Allele Frequency | Inheritance OMIM | Pathogenicity | Co-segregation (Amount of Further Family Members) | Literature, Submitter |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 001 | m | Caucasian | RP | NR2E3 NM_014249.3 | c.[119-2A>C];[=], c.[1171_1172delTT];[=] | p.?, p.Phe391Profs*15 | 0.0005101 Not found | AD, AR | Pathogenic Pathogenic | 2 | [23] Human Genetics-Radboudumc, Radboudumc |
| 002 | m | German | RP, D.D. Usher | USH2A NM_206933.2 | c.[4714C>T];[=], c.[2299delG];[=] | p.Leu1572Phe, p.Glu767Serfs*21 | 0.0006210 0.0005376 | AR | Likely Benign Pathogenic | No | [24] [25] |
| 003 | m | Romanian | Chloridemia, D.D. RP | USH2A NM_206933.2 | c.[11864G>A];[=], c.[2299delG];[=], c.[4714C>T];[=] | p.Trp3955*, p.Glu767Serfs*21, p.Leu1572Phe | 0.0001187 0.0005376 0.0006210 | AR | Pathogenic Pathogenic Likely Benign | 2 | [24,25,26] |
| 004 | f | German | Usher | USH2A NM_206933.2 | c.[10561T>C];[=], c.[4714C>T];[=], c.[2299del];[=] | p.Trp3521Arg, p.Leu1572Phe, p.Glu767Serfs*21 | 0.00006282 0.0006210 0.0005376 | AR | Pathogenic Likely Benign Pathogenic | 9 | [27] [24] [25] |
| 005 | f | Caucasian | CRD | C2orf71 NM_001029883.2 | c.[1709_1728del];[=], c.[2655delT];[=] | p.Gly570Glufs*3, p.Ser885Serfs*2 | 0.000004009 Not found | _ | Pathogenic Pathogenic | 2 | [13] Novel |
| 006 | m | German | CD/CRD | RP1L1 NM_178857.6 | c.[3514C>A];[=], c.[130C>G];[=] | p.Leu1172Ile, p.Pro44Ala | 0.002793 0.01064 | AD, AR | Likely benign Likely benign | No | [28] Illumina Clinical Services Laboratory, Illumina |
| 007 | f | German | CD | RP1L1 NM_178857.6 | c.[133C>T];[=] | p.Arg45Trp | 0.00002028 | AD, AR | Pathogenic | 1 | [29] |
| 008 | m | Caucasian | CRD | RIMS1 NM_001168407.1 | c.[1919C>A];[=] | p.Ser640Tyr | Not found | _ | VUS | No | Novel |
| 009 | f | German | RP | PDE6A NM_000440.2 | c.[607T>A];[=] | p.Phe203Ile | 0.000007953 | _ | VUS | No | Novel |
| 010 | f | Bosnian and Herzegovinan | RP | RP1 NM_006269.1 | c.[5957G>A];[=] | p.Gly1986Asp | Not found | AD, AR | VUS | No | Novel |
| 011 | f | Caucasian | RP | MAK NM_001242957.3 | c.[37G>A];[=], c.[1465+2dupT];[=] | p.Gly13Ser, p.? | 0.000007955 Not found | AR | Pathogenic Pathogenic | 1 | [30] Novel |
| 012 | m | German/Spanish | CD | GUCA1A NM_000409.4 | c.[451C>T];[=] | p.Leu151Phe | Not found | AD | Pathogenic | No | [31] |
| 013 | m | German | MD | PROM1 NM_006017 | c.[1117C>T];[=] | p.Arg373Cys | Not found | AD, AR | Pathogenic | No | [32] |
| 014 | f | Caucasian | RP | PROM1 NM_006017 | c.[1069G>C];[=] | p.Val357Leu | Not found | AD, AR | Benign | 2 | Novel |
| 015 | f | German | Usher | MYO7A NM000260.4 USH2A NM_206933.2 | MYO7A c.[1556G>A];[=], c.[3602G>A];[=] USH2A c.[6883G>A];[=] | MYO7A p.Gly519Asp, p.Cys1201Tyr USH2A p.Gly2295Arg | 0.00001205 0.000004014 0.00002095 | AD, AR; AR | Pathogenic VUS VUS | No | [33] CeGaT Praxis fuer Humangenetik Tuebingen CeGaT Praxis fuer Humangenetik Tuebingen |
| 016 | f | Caucasian | RP, duplex kidney with hypertrophy | SNRNP200 NM_014014.4 | c.[3260C>T];[=] | p.Ser1087Leu | 0.000003977 | AD | Pathogenic | No | [34] |
| 017 | f | Caucasian | RP | RPE65 NM_000329.2 | c.[1154C>T];[=] | p.Thr385Met | 0.0002449 | AD; AR | VUS | No | Illumina Clinical Services Laboratory, Illumina |
| 018 | f | German | RP | PRPF8, NM_006445.3 | c.[1098+6del];[=] | p.? | Not found | AD | VUS | No | Novel |
| 019 | f | Ukrainian | STGD | CTNNA1 NM_001903.4 | c.[1310C>T];[=] | p.(Ala437Val) | 0.0005064 | AD | VUS | No | Ambry Genetics |
| 020 | f | German | MD | OFD1 NM_003611.2 | c.[74A>G];[=] | p.Gln25Arg | 0.000005449 | XLD | VUS | No | Novel |
| 021 | f | Caucasian | STGD | ABCA4 NM_000350.2 | c.[3113C>T];[=], c.[1622T>C];[=] | p.Ala1038Val, p.Leu541Pro | 0.001755 0.0001627 | AR | Pathogenic Pathogenic | No | [35,36] |
| 022 | m | Polish/Caucasian | STGD | ABCA4 NM_000350.2 | c.[5882G>A];[=], c.[3113C>T];[=], c.[1622T>C];[=], c.[6006-5T>G];[=] | p.Gly1961Glu, p.Ala1038Val, p.Leu541Pro, p.? | 0.004564 0.001755 0.0001627 Not found | AR | Pathogenic Pathogenic Pathogenic VUS | 1 | [35,36,37] Novel |
| 023 | m | Caucasian | STGD | ABCA4 NM_000350.2 | c.[4539+1770C>A];[=], c.[5196+1015A>G];[=] | p.?, p.? | Not found Not found | AR | VUS VUS | No | [19] |
| 024 | m | Caucasian | STGD | ABCA4 NM_000350.2 | c.[6601_6602delAG];[=], c.[4253+43G>A];[=] | p.Arg2201fs* p.Ile1377Hisfs*3 | Not found 0.004694 | AR | Pathogenic Pathogenic | 2 | [36] [19] |
| 025 | m | German/Caucasian | STGD | ABCA4 NM_000350.2 | c.[3113C>T];[=], c.[6118C>T];[=] | p.Ala1038Val, p.Arg2040* | 0.001755 0.00001415 | AR | Pathogenic Pathogenic | 2 | [35] EGL Genetic Diagnostics |
| 026 | m | Afghan | STGD | ABCA4 NM_000350.2 | c.[5882G>A];[ 5882G>A], c.[123G>A];[=] | p.Gly1961Glu, p.Trp41* | 0.004564 0.000003978 | AR | Pathogenic Pathogenic | No | [37] EGL Genetic Diagnostics |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, F.; Owczarek-Lipska, M.; Ahmels, T.; Book, M.; Aisenbrey, S.; Menghini, M.; Barthelmes, D.; Schrader, S.; Spital, G.; Neidhardt, J. High-Throughput Sequencing to Identify Mutations Associated with Retinal Dystrophies. Genes 2021, 12, 1269. https://doi.org/10.3390/genes12081269
Song F, Owczarek-Lipska M, Ahmels T, Book M, Aisenbrey S, Menghini M, Barthelmes D, Schrader S, Spital G, Neidhardt J. High-Throughput Sequencing to Identify Mutations Associated with Retinal Dystrophies. Genes. 2021; 12(8):1269. https://doi.org/10.3390/genes12081269
Chicago/Turabian StyleSong, Fei, Marta Owczarek-Lipska, Tim Ahmels, Marius Book, Sabine Aisenbrey, Moreno Menghini, Daniel Barthelmes, Stefan Schrader, Georg Spital, and John Neidhardt. 2021. "High-Throughput Sequencing to Identify Mutations Associated with Retinal Dystrophies" Genes 12, no. 8: 1269. https://doi.org/10.3390/genes12081269
APA StyleSong, F., Owczarek-Lipska, M., Ahmels, T., Book, M., Aisenbrey, S., Menghini, M., Barthelmes, D., Schrader, S., Spital, G., & Neidhardt, J. (2021). High-Throughput Sequencing to Identify Mutations Associated with Retinal Dystrophies. Genes, 12(8), 1269. https://doi.org/10.3390/genes12081269