
Population Genomics Reveals Gene Flow and Adaptive Signature in Invasive Weed Mikania micrantha

Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material and Soil Sampling
2.2. DNA Extraction and GBS Library Sequencing
2.3. Data Quality Control and SNP Calling
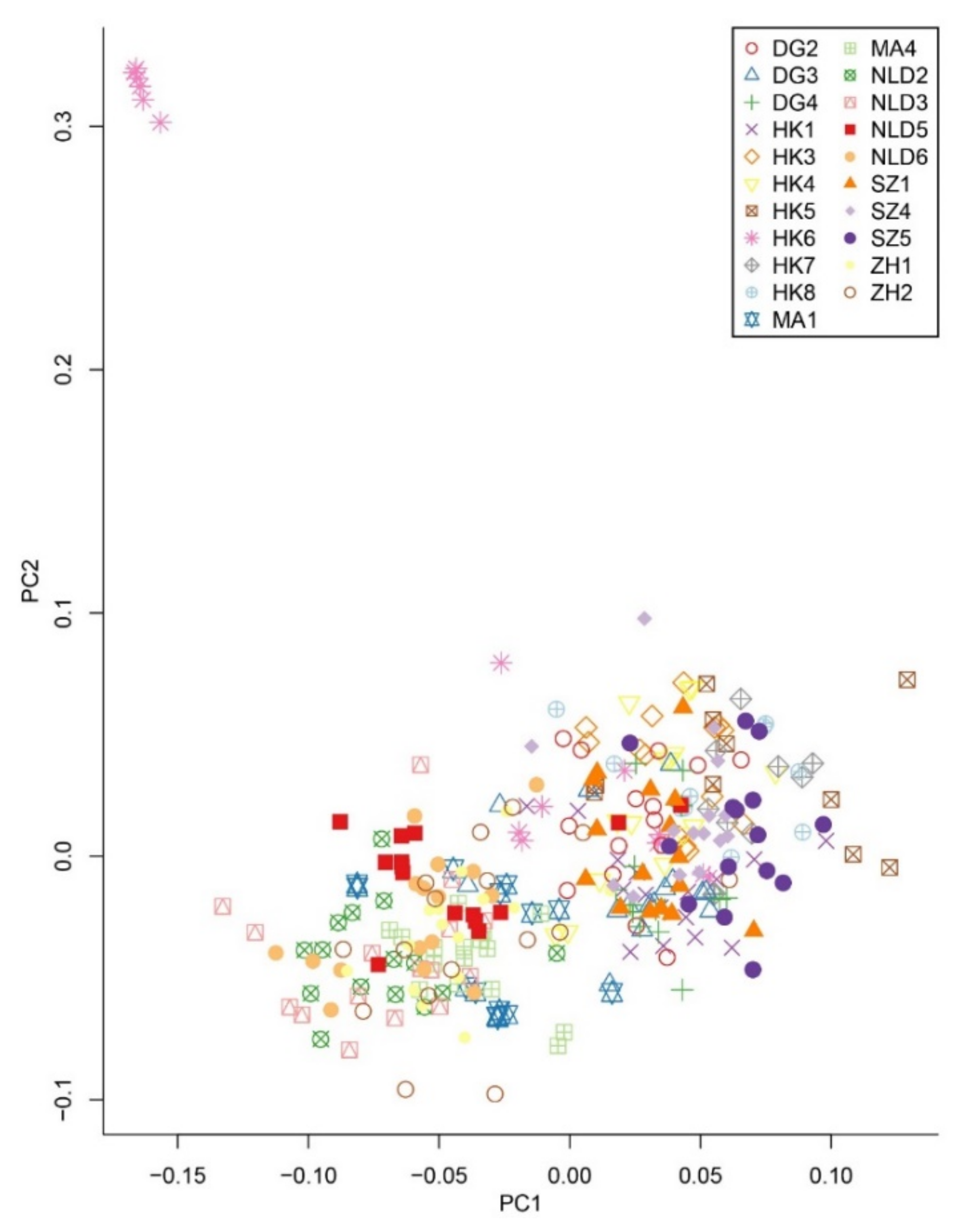
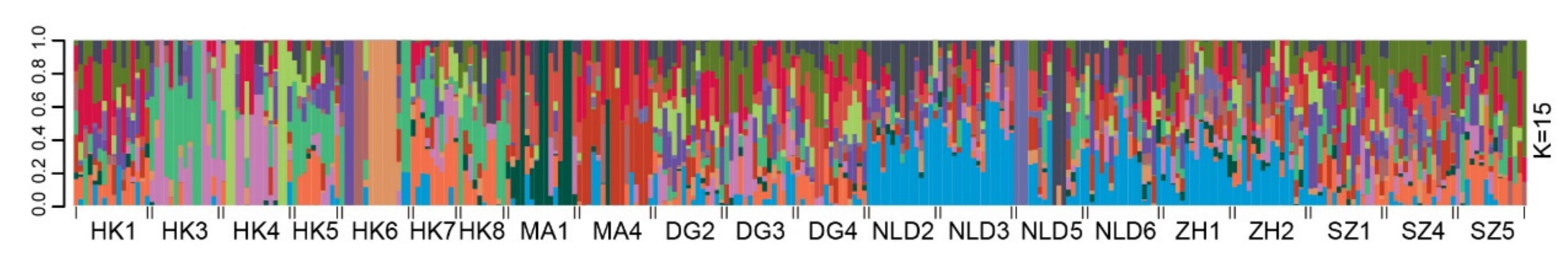
2.4. Genetic Variation and Population Structure
2.5. Environmental Variables
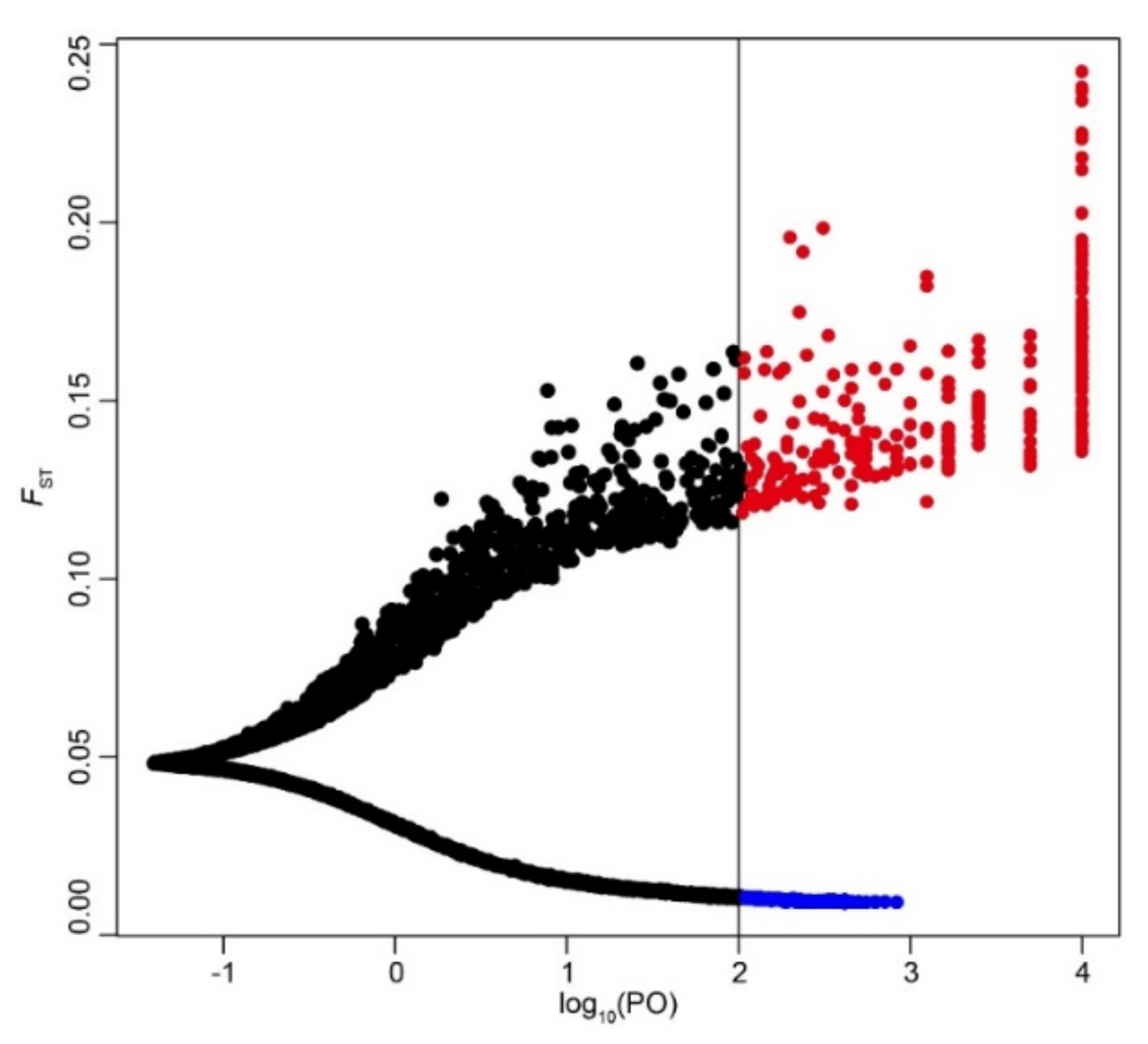
2.6. Identification of Candidate Selective Loci and Function Annotation
2.7. Association of Candidate Selective Loci with Environmental Variables
2.8. Gene Family Analysis
2.9. Identification of Positively Selected Genes
3. Results
3.1. GBS Sequencing and SNP Calling
3.2. Genetic Variation and Population Genetic Structure
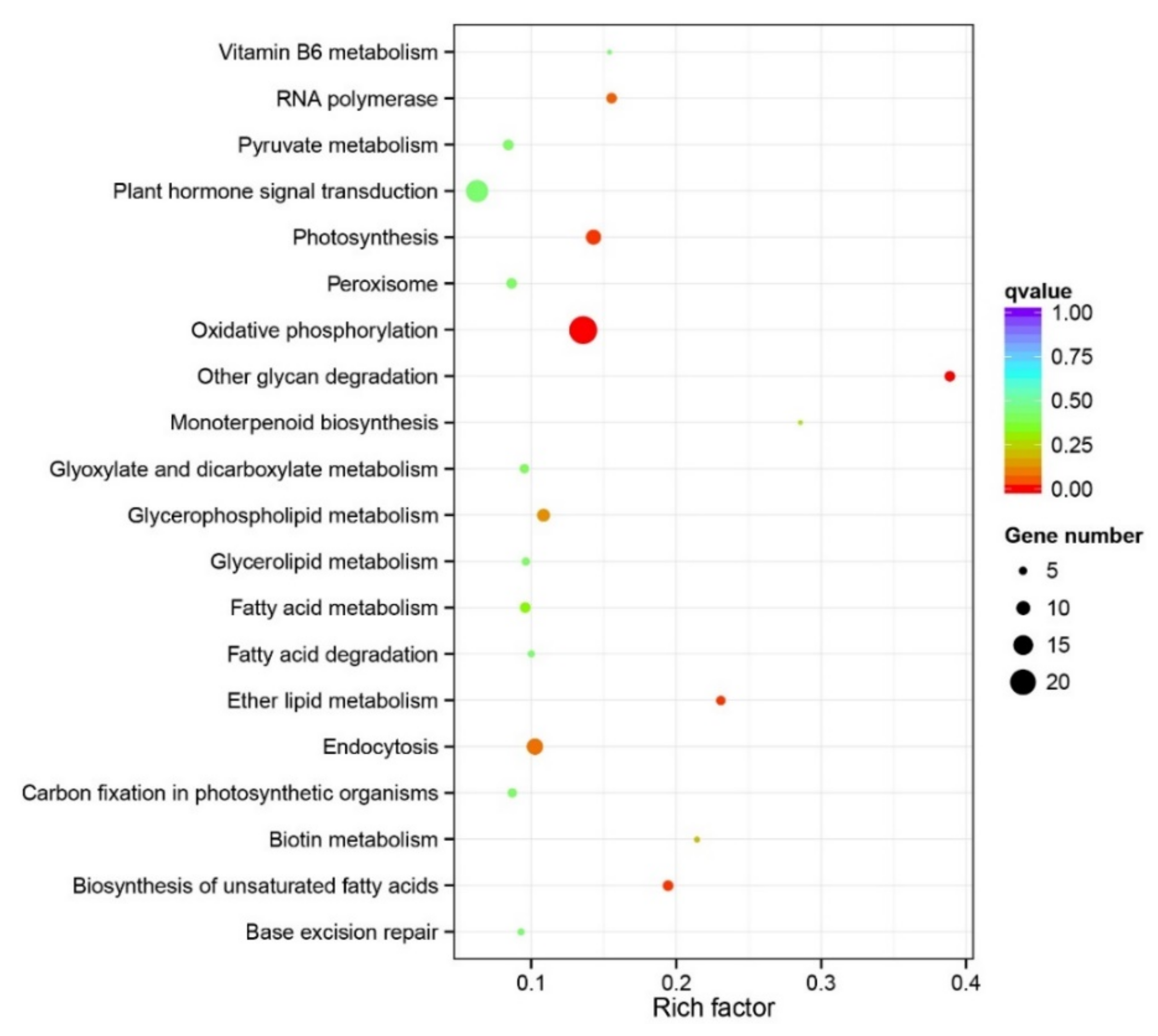
3.3. Identification of Candidate Selective Loci and Gene Annotation
3.4. Association of Candidate Selective Loci with Environmental Variables
3.5. Gene Family Analysis
3.6. Positive Selection Gene Analysis
4. Discussion
4.1. Population Variation and Structure
4.2. Adaptive Response to Environmental Variables
4.3. Genes Unique to M. micrantha May Be Important for Adaptation
4.4. Role of Adaptive Genes in M. micrantha
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Seebens, H.; Blackburn, T.M.; Dyer, E.E.; Genovesi, P.; Hulme, P.E.; Jeschke, J.M.; Pagad, S.; Pysek, P.; Winter, M.; Arianoutsou, M.; et al. No saturation in the accumulation of alien species worldwide. Nat. Commun. 2017, 8, 14435. [Google Scholar] [CrossRef] [PubMed]
- Briski, E.; Chan, F.T.; Darling, J.A.; Lauringson, V.; MacIsaac, H.J.; Zhan, A.; Bailey, S.A. Beyond propagule pressure: Importance of selection during the transport stage of biological invasions. Front. Ecol. Environ. 2018, 16, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Bock, D.G.; Caseys, C.; Cousens, R.D.; Hahn, M.A.; Heredia, S.M.; Hubner, S.; Turner, K.G.; Whitney, K.D.; Rieseberg, L.H. What we still don’t know about invasion genetics. Mol. Ecol. 2015, 24, 2277–2297. [Google Scholar] [CrossRef]
- Van Boheemen, L.A.; Lombaert, E.; Nurkowski, K.A.; Gauffre, B.; Rieseberg, L.H.; Hodgins, K.A. Multiple introductions, admixture and bridgehead invasion characterize the introduction history of Ambrosia artemisiifolia in Europe and Australia. Mol. Ecol. 2017, 26, 5421–5434. [Google Scholar] [CrossRef] [PubMed]
- Brandes, U.; Furevik, B.B.; Nielsen, L.R.; Kjær, E.D.; Rosef, L.; Fjellheim, S.; Zhan, A. Introduction history and population genetics of intracontinental scotch broom (Cytisus scoparius) invasion. Divers. Distrib. 2019, 25, 1773–1786. [Google Scholar] [CrossRef] [Green Version]
- Smith, A.L.; Hodkinson, T.R.; Villellas, J.; Catford, J.A.; Csergo, A.M.; Blomberg, S.P.; Crone, E.E.; Ehrlen, J.; Garcia, M.B.; Laine, A.-L.; et al. Global gene flow releases invasive plants from environmental constraints on genetic diversity. Proc. Natl. Acad. Sci. USA 2020, 117, 4218–4227. [Google Scholar] [CrossRef] [Green Version]
- Eckert, C.G.; Samis, K.E.; Lougheed, S.C. Genetic variation across species’ geographical ranges: The central-marginal hypothesis and beyond. Mol. Ecol. 2008, 17, 1170–1188. [Google Scholar] [CrossRef]
- Keller, S.R.; Taylor, D.R. Genomic admixture increases fitness during a biological invasion. J. Evol. Biol. 2010, 23, 1720–1731. [Google Scholar] [CrossRef]
- Qiao, H.; Liu, W.; Zhang, Y.; Zhang, Y.-Y.; Li, Q.Q. Genetic admixture accelerates invasion via provisioning rapid adaptive evolution. Mol. Ecol. 2019, 28, 4012–4027. [Google Scholar] [CrossRef]
- Prentis, P.J.; Wilson, J.R.U.; Dormontt, E.E.; Richardson, D.M.; Lowe, A.J. Adaptive evolution in invasive species. Trends Plant Sci. 2008, 13, 288–294. [Google Scholar] [CrossRef]
- Lee, C.E. Evolutionary genetics of invasive species. Trends Ecol. Evol. 2002, 17, 386–391. [Google Scholar] [CrossRef]
- Moran, E.V.; Alexander, J.M. Evolutionary responses to global change: Lessons from invasive species. Ecol. Lett. 2014, 17, 637–649. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Liu, Z.; Pan, Q.; Chen, X.; Wang, H.; Guo, H.; Liu, S.; Lu, H.; Tian, S.; Li, R.; et al. Genomic analyses reveal demographic history and temperate adaptation of the newly discovered honey bee subspecies Apis mellifera sinisxinyuan n. ssp. Mol. Biol. Evol. 2016, 33, 1337–1348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodgins, K.A.; Lai, Z.; Nurkowski, K.; Huang, J.; Rieseberg, L.H. The molecular basis of invasiveness: Differences in gene expression of native and introduced common ragweed (Ambrosia artemisiifolia) in stressful and benign environments. Mol. Ecol. 2013, 22, 2496–2510. [Google Scholar] [CrossRef]
- Van Boheemen, L.A.; Hodgins, K.A. Rapid repeatable phenotypic and genomic adaptation following multiple introductions. Mol. Ecol. 2020, 29, 4102–4117. [Google Scholar] [CrossRef]
- Davey, J.W.; Hohenlohe, P.A.; Etter, P.D.; Boone, J.Q.; Catchen, J.M.; Blaxter, M.L. Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nat. Rev. Genet. 2011, 12, 499–510. [Google Scholar] [CrossRef]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef] [Green Version]
- Martin, M.D.; Olsen, M.T.; Samaniego, J.A.; Zimmer, E.A.; Gilbert, M.T.P. The population genomic basis of geographic differentiation in North American common ragweed (Ambrosia artemisiifolia L.). Ecol. Evol. 2016, 6, 3760–3771. [Google Scholar] [CrossRef] [PubMed]
- Holm, L.G.; Plucknett, D.L.; Pancho, J.V.; Herberger, J.P. The World’s Worst Weeds; University Press of Hawaii: Honolulu, HI, USA, 1977. [Google Scholar]
- Bhatt, J.R.; Singh, J.S.; Singh, S.P.; Tripathi, R.S.; Kohli, R.K. Biology of Mikania micrantha H.B.K.: A Review. In Invasive Alien Plants: An Ecological Appraisal for the Indian Subcontinent; CAB International: Oxford, UK, 2012; pp. 99–107. [Google Scholar]
- Zhang, L.Y.; Ye, W.H.; Cao, H.L.; Feng, H.L. Mikania micrantha H.B.K. in China—An overview. Weed Res. 2004, 44, 42–49. [Google Scholar] [CrossRef]
- Dong, L.; Wu, L.F. New research progress of Mikania micrantha H.B.K. J. Anhui Agric. Sci. 2011, 39, 15352–15355. [Google Scholar]
- Banerjee, A.K.; Mukherjee, A.; Dewanji, A. Potential distribution of Mikania micrantha Kunth in India—Evidence of climatic niche and biome shifts. Flora 2017, 234, 215–223. [Google Scholar] [CrossRef]
- Yue, M.; Yu, H.; Li, W.; Yin, A.; Cui, Y.; Tian, X. Flooding with shallow water promotes the invasiveness of Mikania micrantha. Ecol. Evol. 2019, 9, 9177–9184. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, A.K.; Mukherjee, A.; Guo, W.; Liu, Y.; Huang, Y. Spatio-temporal patterns of climatic niche dynamics of an invasive plant Mikania micrantha Kunth and its potential distribution under projected climate change. Front. Ecol. Evol. 2019, 7, 291. [Google Scholar] [CrossRef] [Green Version]
- Deng, X. Morphological and physiological plasticity responding to different light environments of the invasive plant, Mikania micrantha H.B.Kunth. Ecol. Environ. Sci. 2010, 19, 1170–1175. [Google Scholar]
- Banerjee, A.K.; Mukherjee, A.; Guo, W.; Ng, W.L.; Huang, Y. Combining ecological niche modeling with genetic lineage information to predict potential distribution of Mikania micrantha Kunth in South and Southeast Asia under predicted climate change. Glob. Ecol. Conserv. 2019, 20, e00800. [Google Scholar] [CrossRef]
- Guo, Q.; Qiang, S.; Lin, J.; Yu, Y. The biological characteristics and integrated management of Mikania micrantha. Wuyi Sci. J. 2005, 21, 72–76. [Google Scholar]
- Wang, B.; Liao, W.; Zan, Q.; Li, M.; Zhou, X.; Gao, S. The spreads of Mikania micrantha in China. Acta Sci. Nat. Univ. Sunyatseni 2003, 42, 47–50. [Google Scholar]
- Wang, T.; Su, Y.; Chen, G. Population genetic variation and structure of the invasive weed Mikania micrantha in southern China: Consequences of rapid range expansion. J. Hered. 2008, 99, 22–33. [Google Scholar] [CrossRef]
- Wang, T.; Chen, G.; Zan, Q.; Wang, C.; Su, Y. AFLP genome scan to detect genetic structure and candidate loci under selection for local adaptation of the invasive weed Mikania micrantha. PLoS ONE 2012, 7, e41310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geng, S.L.; Chen, Q.; Cai, W.L.; Cao, A.C.; Ou-Yang, C.B. Genetic variation in the invasive weed Mikania micrantha (Asteraceae) suggests highways as corridors for its dispersal in southern China. Ann. Bot. 2017, 119, 457–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banerjee, A.K.; Hou, Z.; Lin, Y.; Lan, W.; Tan, F.; Xing, F.; Li, G.; Guo, W.; Huang, Y. Going with the flow: Analysis of population structure reveals high gene flow shaping invasion pattern and inducing range expansion of Mikania micrantha in Asia. Ann. Bot. 2020, 125, 1113–1126. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Wang, Z.; Chen, G.; Wang, C.; Su, Y. Invasive chloroplast population genetics of Mikania micrantha in China: No local adaptation and negative correlation between diversity and geographic distance. Front. Plant Sci. 2016, 7, 1426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, J.; Wang, Z.; Su, Y.; Wang, T. Associations between population epigenetic differentiation and environmental factors in the exotic weed mile-a-minute (Mikania micrantha). Weed Sci. 2021, 69, 307–332. [Google Scholar] [CrossRef]
- Su, Y.J.; Wang, T.; Zheng, B.; Jiang, Y.; Chen, G.P.; Ouyang, P.Y.; Sun, Y.F. Genetic differentiation of relictual populations of Alsophila spinulosa in southern China inferred from cpDNA trnL-F noncoding sequences. Mol. Phylogenet. Evol. 2005, 34, 323–333. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Subgroup, G.P.D.P. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Goudet, J. Hierfstat, a package for R to compute and test hierarchical F-statistics. Mol. Ecol. Notes 2005, 5, 184–186. [Google Scholar] [CrossRef] [Green Version]
- Pohlert, T. The Pairwise Multiple Comparison of Mean Ranks Package (PMCMR). Available online: https://cran.r-project.org/web/packages/PMCMR/index.html (accessed on 6 March 2018).
- Kamvar, Z.N.; Tabima, J.F.; Grunwald, N.J. Poppr: An R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ 2014, 2, e281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keenan, K.; McGinnity, P.; Cross, T.F.; Crozier, W.W.; Prodöhl, P.A. diveRsity: An R package for the estimation and exploration of population genetics parameters and their associated errors. Methods Ecol. Evol. 2013, 4, 782–788. [Google Scholar] [CrossRef] [Green Version]
- Stéphane, D.; Anne-Béatrice, D. The ade4 package: Implementing the duality diagram for ecologists. J. Stat. Softw. 2007, 22, 1–20. [Google Scholar]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexander, D.H.; Lange, K. Enhancements to the ADMIXTURE algorithm for individual ancestry estimation. BMC Bioinform. 2011, 12, 246. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011, 88, 76–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A. FigTree v1.4.2: Tree Figure Drawing Tool. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 5 March 2019).
- Huete, A.; Didan, K.; Miura, T.; Rodriguez, E.P.; Gao, X.; Ferreira, L.G. Overview of the radiometric and biophysical performance of the MODIS vegetation indices. Remote Sens. Environ. 2002, 83, 195–213. [Google Scholar] [CrossRef]
- Naimi, B.; Hamm, N.A.S.; Groen, T.A.; Skidmore, A.K.; Toxopeus, A.G. Where is positional uncertainty a problem for species distribution modelling? Ecography 2014, 37, 191–203. [Google Scholar] [CrossRef]
- Foll, M.; Gaggiotti, O. A genome-scan method to identify selected loci appropriate for both dominant and codominant markers: A Bayesian perspective. Genetics 2008, 180, 977–993. [Google Scholar] [CrossRef] [Green Version]
- Lotterhos, K.E.; Whitlock, M.C. Evaluation of demographic history and neutral parameterization on the performance of FST outlier tests. Mol. Ecol. 2014, 23, 2178–2192. [Google Scholar] [CrossRef] [Green Version]
- Eddy, S.R. Profile hidden Markov models. Bioinformatics 1998, 14, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Frichot, E.; François, O. LEA: An R package for landscape and ecological association studies. Methods Ecol. Evol. 2015, 6, 925–929. [Google Scholar] [CrossRef]
- Frichot, E.; Schoville, S.D.; Bouchard, G.; Francois, O. Testing for associations between loci and environmental gradients using latent factor mixed models. Mol. Biol. Evol. 2013, 30, 1687–1699. [Google Scholar] [CrossRef] [Green Version]
- Günther, T.; Coop, G. A short manual for BayEnv2. Available online: https://bitbucket.org/tguenther/bayenv2_public/src/default/bayenv2_manual.pdf (accessed on 19 March 2021).
- Li, L.; Stoeckert, C.J., Jr.; Roos, D.S. OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar] [CrossRef] [Green Version]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [Green Version]
- Lucardi, R.D.; Wallace, L.E.; Ervin, G.N. Invasion success in cogongrass (Imperata cylindrica): A population genetic approach exploring genetic diversity and historical iIntroductions. Invasive Plant Sci. Manag. 2017, 7, 59–75. [Google Scholar] [CrossRef]
- Kalb, D.M.; Delaney, D.A.; DeYoung, R.W.; Bowman, J.L. Genetic diversity and demographic history of introduced sika deer on the Delmarva Peninsula. Ecol. Evol. 2019, 9, 11504–11517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allendorf, F.W. Genetic drift and the loss of alleles versus heterozygosity. Zoo Biol. 1986, 5, 181–190. [Google Scholar] [CrossRef]
- Xia, L.; Geng, Q.; An, S. Rapid genetic divergence of an invasive species, Spartina alterniflora, in China. Front. Genet. 2020, 11, 284. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; He, Z.; Huang, Y.; Lu, L.; Yan, Y.; Hong, L.; Shen, H.; Liu, Y.; Guo, Q.; Jiang, L.; et al. The emergence of the hyperinvasive vine, Mikania micrantha (Asteraceae), via admixture and founder events inferred from population transcriptomics. Mol. Ecol. 2017, 26, 3405–3423. [Google Scholar] [CrossRef] [PubMed]
- Chun, Y.J.; Fumanal, B.; Laitung, B.; Bretagnolle, F. Gene flow and population admixture as the primary post-invasion processes in common ragweed (Ambrosia artemisiifolia) populations in France. New Phytol. 2010, 185, 1100–1107. [Google Scholar] [CrossRef] [PubMed]
- Hong, L.; Shen, H.; Ye, W.H.; Cao, H.L.; Wang, Z.M. Self-incompatibility in Mikania micrantha in South China. Weed Res. 2007, 47, 280–283. [Google Scholar] [CrossRef]
- Li, M.; Lu, E.; Guo, Q.; Zan, Q.; Wei, P.; Jiang, L.; Xu, H.; Zhong, T. Evaluation of the controlling methods and strategies for Mikania micrantha H.B.K. Acta Ecol. Sinica 2012, 32, 3240–3251. [Google Scholar]
- Guo, Q.F. Central-marginal population dynamics in species invasions. Front. Ecol. Evol. 2014, 2, 23. [Google Scholar] [CrossRef] [Green Version]
- Guo, Q.; Taper, M.; Schoeneberger, M.M.; Brandle, J.R. Spatial temporal population dynamics across a species’ range: From center to margin. Oikos 2005, 108, 47–57. [Google Scholar] [CrossRef] [Green Version]
- Bravo-Monzón, Á.E.; González-Rodríguez, A.; Espinosa-García, F.J. Spatial structure of genetic and chemical variation in native populations of the mile-a-minute weed Mikania micrantha. Biochem. Syst. Ecol. 2018, 76, 23–31. [Google Scholar] [CrossRef]
- Winkler, D.E.; Chapin, K.J.; Francois, O.; Garmon, J.D.; Gaut, B.S.; Huxman, T.E. Multiple introductions and population structure during the rapid expansion of the invasive Sahara mustard (Brassica tournefortii). Ecol. Evol. 2019, 9, 7928–7941. [Google Scholar] [CrossRef] [Green Version]
- Vanden Broeck, A.; Van Landuyt, W.; Cox, K.; De Bruyn, L.; Gyselings, R.; Oostermeijer, G.; Valentin, B.; Bozic, G.; Dolinar, B.; Illyés, Z.; et al. High levels of effective long-distance dispersal may blur ecotypic divergence in a rare terrestrial orchid. BMC Ecol. 2014, 14, 20. [Google Scholar] [CrossRef] [Green Version]
- Choudhury, M.R.; Deb, P.; Singha, H.; Chakdar, B.; Medhi, M. Predicting the probable distribution and threat of invasive Mimosa diplotricha Suavalle and Mikania micrantha Kunth in a protected tropical grassland. Ecol. Eng. 2016, 97, 23–31. [Google Scholar] [CrossRef]
- Chen, B.; Su, J.; Liao, H.; Peng, S. A greater foraging scale, not a higher foraging precision, may facilitate invasion by exotic plants in nutrient-heterogeneous conditions. Ann. Bot. 2018, 121, 561–569. [Google Scholar] [CrossRef] [PubMed]
- Stitt, M.; Gibon, Y.; Lunn, J.E.; Piques, M. Multilevel genomics analysis of carbon signalling during low carbon availability: Coordinating the supply and utilisation of carbon in a fluctuating environment. Funct. Plant Biol. 2007, 34, 526–549. [Google Scholar] [CrossRef]
- McLaughlin, J.E.; Boyer, J.S. Glucose localization in maize ovaries when kernel number decreases at low water potential and sucrose is fed to the stems. Ann. Bot. 2004, 94, 75–86. [Google Scholar] [CrossRef] [Green Version]
- Valmonte, G.R.; Arthur, K.; Higgins, C.M.; MacDiarmid, R.M. Calcium-dependent protein kinases in plants: Evolution, expression and function. Plant Cell Physiol. 2014, 55, 551–569. [Google Scholar] [CrossRef] [Green Version]
- Hafsi, C.; Falleh, H.; Saada, M.; Ksouri, R.; Abdelly, C. Potassium deficiency alters growth, photosynthetic performance, secondary metabolites content, and related antioxidant capacity in Sulla carnosa grown under moderate salinity. Plant Physiol. Bioch. 2017, 118, 609–617. [Google Scholar] [CrossRef]
- Sardans, J.; Peñuelas, J. Potassium: A neglected nutrient in global change. Glob. Ecol. Biogeogr. 2015, 24, 261–275. [Google Scholar] [CrossRef] [Green Version]
- Ou, Q.; Yang, Y.; Liang, W.; Sun, F.; Peng, C. Effects of leaf leachates of the invasive plant Mikania micrantha H.B.K. on soil potassium activation and soil enzyme activity. J. South China Norm. Univ. (Nat. Sci. Ed.) 2020, 52, 63–69. [Google Scholar]
- Cooke, J.; Leishman, M.R. Consistent alleviation of abiotic stress with silicon addition: A meta-analysis. Funct. Ecol. 2016, 30, 1340–1357. [Google Scholar] [CrossRef]
- Zhou, Y.; Su, Y.; Zhong, Y.; Xie, P.; Xu, M.; Su, Z. Community attributes predict the relationship between habitat invasibility and land use types in an agricultural and forest landscape. Forests 2019, 10, 867. [Google Scholar] [CrossRef] [Green Version]
- Shen, Q.; Zhang, L.; Liao, Z.; Wang, S.; Yan, T.; Shi, P.; Liu, M.; Fu, X.; Pan, Q.; Wang, Y.; et al. The genome of Artemisia annua provides insight into the evolution of Asteraceae family and artemisinin biosynthesis. Mol. Plant 2018, 11, 776–788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, C.; Liu, Y.; Song, A.; Dong, G.; Zhao, H.; Sun, W.; Ramakrishnan, S.; Wang, Y.; Wang, S.; Li, T.; et al. The Chrysanthemum nankingense genome provides insights into the evolution and diversification of Chrysanthemum flowers and medicinal traits. Mol. Plant 2018, 11, 1482–1491. [Google Scholar] [CrossRef] [Green Version]
- Tian, D.; Pan, X.; Yu, Y.; Wang, W.; Zhang, F.; Ge, Y.; Shen, X.; Shen, F.; Liu, X. De novo characterization of the Anthurium transcriptome and analysis of its digital gene expression under cold stress. BMC Genom. 2013, 14, 827. [Google Scholar] [CrossRef] [Green Version]
- He, M.; Qin, C.; Wang, X.; Ding, N. Plant unsaturated fatty acids: Biosynthesis and regulation. Front. Plant Sci. 2020, 11, 390. [Google Scholar] [CrossRef] [PubMed]
- Cui, C.; Wang, Z.; Su, Y.; Wang, T. New insight into the rapid growth of the Mikania micrantha stem based on DIA proteomic and RNA-Seq analysis. J. Proteom. 2021, 236, 104126. [Google Scholar] [CrossRef]
- Wu, M.; Li, Z.; Wang, J. Transcriptional analyses reveal the molecular mechanism governing shade tolerance in the invasive plant Solidago canadensis. Ecol. Evol. 2020, 10, 4391–4406. [Google Scholar] [CrossRef] [Green Version]
- Azzouz-Olden, F.; Hunt, A.G.; Dinkins, R. Transcriptome analysis of drought-tolerant sorghum genotype SC56 in response to water stress reveals an oxidative stress defense strategy. Mol. Biol. Rep. 2020, 47, 3291–3303. [Google Scholar] [CrossRef] [Green Version]
- Peters, L.P.; Carvalho, G.; Vilhena, M.B.; Creste, S.; Azevedo, R.A.; Monteiro-Vitorello, C.B. Functional analysis of oxidative burst in sugarcane smut-resistant and -susceptible genotypes. Planta 2017, 245, 749–764. [Google Scholar] [CrossRef] [Green Version]
- Shin, S.; Zheng, P.; Fazio, G.; Mazzola, M.; Main, D.; Zhu, Y. Transcriptome changes specifically associated with apple (Malus domestica) root defense response during Pythium ultimum infection. Physiol. Mol. Plant Pathol. 2016, 94, 16–26. [Google Scholar] [CrossRef] [Green Version]
- Qiu, Q.; Zhang, G.; Ma, T.; Qian, W.; Wang, J.; Ye, Z.; Cao, C.; Hu, Q.; Kim, J.; Larkin, D.M.; et al. The yak genome and adaptation to life at high altitude. Nat. Genet. 2012, 44, 946–951. [Google Scholar] [CrossRef] [Green Version]
- Zou, C.; Yu, D. Analysis of the cold-responsive transcriptome in the mature pollen of Arabidopsis. J. Plant Biol. 2010, 53, 400–416. [Google Scholar]
- Ribeiro, P.R.; Willems, L.A.J.; Silva, A.T.; Fernandez, L.G.; de Castro, R.D.; Bucher, J.; Snoek, B.L.; Hilhorst, H.W.M.; Ligterink, W. Transcriptome profiling of Ricinus communis L. provides new insights underlying the mechanisms towards thermotolerance during seed imbibition and germination. Ind. Crops Prod. 2018, 126, 380–393. [Google Scholar] [CrossRef] [Green Version]
- Dang, H.Q.; Tran, N.Q.; Gill, S.S.; Tuteja, R.; Tuteja, N. A single subunit MCM6 from pea promotes salinity stress tolerance without affecting yield. Plant Mol. Biol. 2011, 76, 19–34. [Google Scholar] [CrossRef]
- Casati, P.; Walbot, V. Maize lines expressing RNAi to chromatin remodeling factors are similarly hypersensitive to UV-B radiation but exhibit distinct transcriptome responses. Epigenetics 2008, 3, 216–229. [Google Scholar] [CrossRef] [Green Version]
- Goellner, E.M.; Putnam, C.D.; Kolodner, R.D. Exonuclease 1-dependent and independent mismatch repair. DNA Repair 2015, 32, 24–32. [Google Scholar] [CrossRef] [Green Version]
- Huang, G.; Ma, S.; Bai, L.; Zhang, L.; Ma, H.; Jia, P.; Liu, J.; Zhong, M.; Guo, Z. Signal transduction during cold, salt, and drought stresses in plants. Mol. Biol. Rep. 2012, 39, 969–987. [Google Scholar] [CrossRef]
- Yuan, H.; Zeng, X.; Ling, Z.; Wei, Z.; Wang, Y.; Zhuang, Z.; Xu, Q.; Tang, Y.; Tashi, N. Transcriptome profiles reveal cold acclimation and freezing tolerance of susceptible and tolerant hulless barley genotypes. Acta Physiol. Plant. 2017, 39, 275. [Google Scholar] [CrossRef]
- Luo, L.; Kong, X.; Gao, Z.; Zheng, Y.; Yang, Y.; Li, X.; Yang, D.; Geng, Y.; Yang, Y. Comparative transcriptome analysis reveals ecological adaption of cold tolerance in northward invasion of Alternanthera philoxeroides. BMC Genom. 2020, 21, 532. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Hu, S.; Wang, X.; Liu, H.; Zhou, Y.; Guan, Q. De novo assembly of Amorpha fruticosa L. transcriptome in response to drought stress provides insight into the tolerance mechanisms. PeerJ 2021, 9, e11044. [Google Scholar] [CrossRef] [PubMed]
- Su, W.; Ye, C.; Zhang, Y.; Hao, S.; Li, Q.Q. Identification of putative key genes for coastal environments and cold adaptation in mangrove Kandelia obovata through transcriptome analysis. Sci. Total Environ. 2019, 681, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Shi, Y.; Senthilkumar, H.A.; Qiao, Q.; Wang, Q.; Shen, Y.; Hu, G. Enriched networks ‘nucleoside/nucleotide and ribonucleoside/ribonucleotide metabolic processes’ and ‘response to stimulus’ potentially conferred to drought adaptation of the epiphytic orchid Dendrobium wangliangii. Physiol. Mol. Biol. Plants 2019, 25, 31–45. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Chen, Y.; Lu, L.; Lu, Y.; Li, L. Transcriptome analysis reveals dynamic changes in the gene expression of tobacco seedlings under low potassium stress. J. Genet. 2015, 94, 397–406. [Google Scholar] [CrossRef]
- Nayak, S.S.; Pradhan, S.; Sahoo, D.; Parida, A. De novo transcriptome assembly and analysis of Phragmites karka, an invasive halophyte, to study the mechanism of salinity stress tolerance. Sci. Rep. 2020, 10, 5192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swaminathan, P.; Ohrtman, M.; Carinder, A.; Deuja, A.; Wang, C.; Gaskin, J.; Fennell, A.; Clay, S. Water deficit transcriptomic responses differ in the invasive Tamarix chinensis and T. ramosissima established in the southern and northern United States. Plants 2020, 9, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Population/Region | AR | HO | HS | FIS |
|---|---|---|---|---|
| HK1 | 1.886 (0.003) | 0.335 (0.003) | 0.374 (0.003) | 0.084 (0.006) |
| HK3 | 1.743 (0.005) | 0.293 (0.004) | 0.364 (0.003) | 0.203 (0.007) |
| HK4 | 1.772 (0.005) | 0.280 (0.003) | 0.424 (0.003) | 0.364 (0.006) |
| HK5 | 1.770 (0.005) | 0.309 (0.004) | 0.374 (0.003) | 0.151 (0.007) |
| HK6 | 1.787 (0.005) | 0.297 (0.004) | 0.407 (0.003) | 0.278 (0.007) |
| HK7 | 1.835 (0.005) | 0.379 (0.004) | 0.427 (0.003) | 0.104 (0.007) |
| HK8 | 1.783 (0.005) | 0.335 (0.004) | 0.392 (0.003) | 0.142 (0.007) |
| HK | 1.983 (0.001) | 0.315 (0.003) | 0.411 (0.002) | 0.247 (0.005) |
| SZ1 | 1.860 (0.004) | 0.323 (0.003) | 0.458 (0.003) | 0.298 (0.006) |
| SZ4 | 1.875 (0.004) | 0.362 (0.004) | 0.426 (0.003) | 0.146 (0.006) |
| SZ5 | 1.857 (0.004) | 0.336 (0.004) | 0.371 (0.003) | 0.093 (0.006) |
| SZ | 1.986 (0.001) | 0.340 (0.003) | 0.428 (0.002) | 0.213 (0.005) |
| DG2 | 1.852 (0.004) | 0.317 (0.003) | 0.448 (0.003) | 0.277 (0.006) |
| DG3 | 1.850 (0.004) | 0.336 (0.003) | 0.391 (0.003) | 0.127 (0.006) |
| DG4 | 1.877 (0.004) | 0.387 (0.004) | 0.430 (0.003) | 0.103 (0.007) |
| DG | 1.979 (0.001) | 0.347 (0.003) | 0.429 (0.002) | 0.196 (0.005) |
| NLD2 | 1.786 (0.005) | 0.297 (0.004) | 0.380 (0.003) | 0.217 (0.007) |
| NLD3 | 1.826 (0.004) | 0.325 (0.004) | 0.394 (0.003) | 0.177 (0.006) |
| NLD5 | 1.820 (0.004) | 0.317 (0.004) | 0.397 (0.003) | 0.195 (0.007) |
| NLD6 | 1.842 (0.004) | 0.308 (0.003) | 0.396 (0.003) | 0.208 (0.006) |
| NLD | 1.965 (0.002) | 0.312 (0.003) | 0.396 (0.003) | 0.227 (0.005) |
| ZH1 | 1.796 (0.005) | 0.292 (0.003) | 0.443 (0.003) | 0.339 (0.006) |
| ZH2 | 1.852 (0.004) | 0.330 (0.003) | 0.419 (0.003) | 0.223 (0.006) |
| ZH | 1.952 (0.002) | 0.312 (0.003) | 0.435 (0.003) | 0.297 (0.005) |
| MA1 | 1.790 (0.005) | 0.333 (0.004) | 0.363 (0.003) | 0.091 (0.007) |
| MA4 | 1.811 (0.005) | 0.321 (0.004) | 0.414 (0.003) | 0.223 (0.007) |
| MA | 1.930 (0.003) | 0.327 (0.003) | 0.404 (0.003) | 0.197 (0.006) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruan, X.; Wang, Z.; Su, Y.; Wang, T. Population Genomics Reveals Gene Flow and Adaptive Signature in Invasive Weed Mikania micrantha. Genes 2021, 12, 1279. https://doi.org/10.3390/genes12081279
Ruan X, Wang Z, Su Y, Wang T. Population Genomics Reveals Gene Flow and Adaptive Signature in Invasive Weed Mikania micrantha. Genes. 2021; 12(8):1279. https://doi.org/10.3390/genes12081279
Chicago/Turabian StyleRuan, Xiaoxian, Zhen Wang, Yingjuan Su, and Ting Wang. 2021. "Population Genomics Reveals Gene Flow and Adaptive Signature in Invasive Weed Mikania micrantha" Genes 12, no. 8: 1279. https://doi.org/10.3390/genes12081279
APA StyleRuan, X., Wang, Z., Su, Y., & Wang, T. (2021). Population Genomics Reveals Gene Flow and Adaptive Signature in Invasive Weed Mikania micrantha. Genes, 12(8), 1279. https://doi.org/10.3390/genes12081279