
Single Cell Sequencing Reveals Mechanisms of Persistent Truncus Arteriosus Formation after PDGFRα and PDGFRβ Double Knockout in Cardiac Neural Crest Cells


Abstract
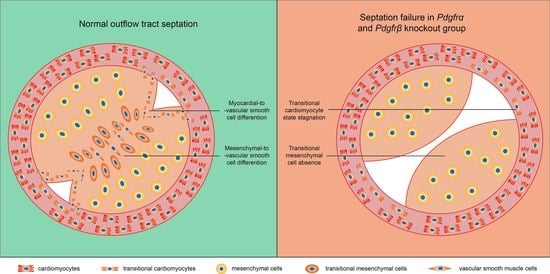
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animal Models
2.2. Embryo Dissection and Single-Cell Library Generation
2.3. Single-Cell Transcriptome Library Preparation and Sequencing
2.4. Processing of Raw Sequencing Reads
2.5. Cell Filtering and Cell-Type Clustering Analysis
2.6. RNA Velocity Analysis
2.7. CellChat Analysis
2.8. Immunofluorescence Experiments
2.9. In Situ Hybridization Experiments
2.10. Statistics and Reproducibility
3. Results
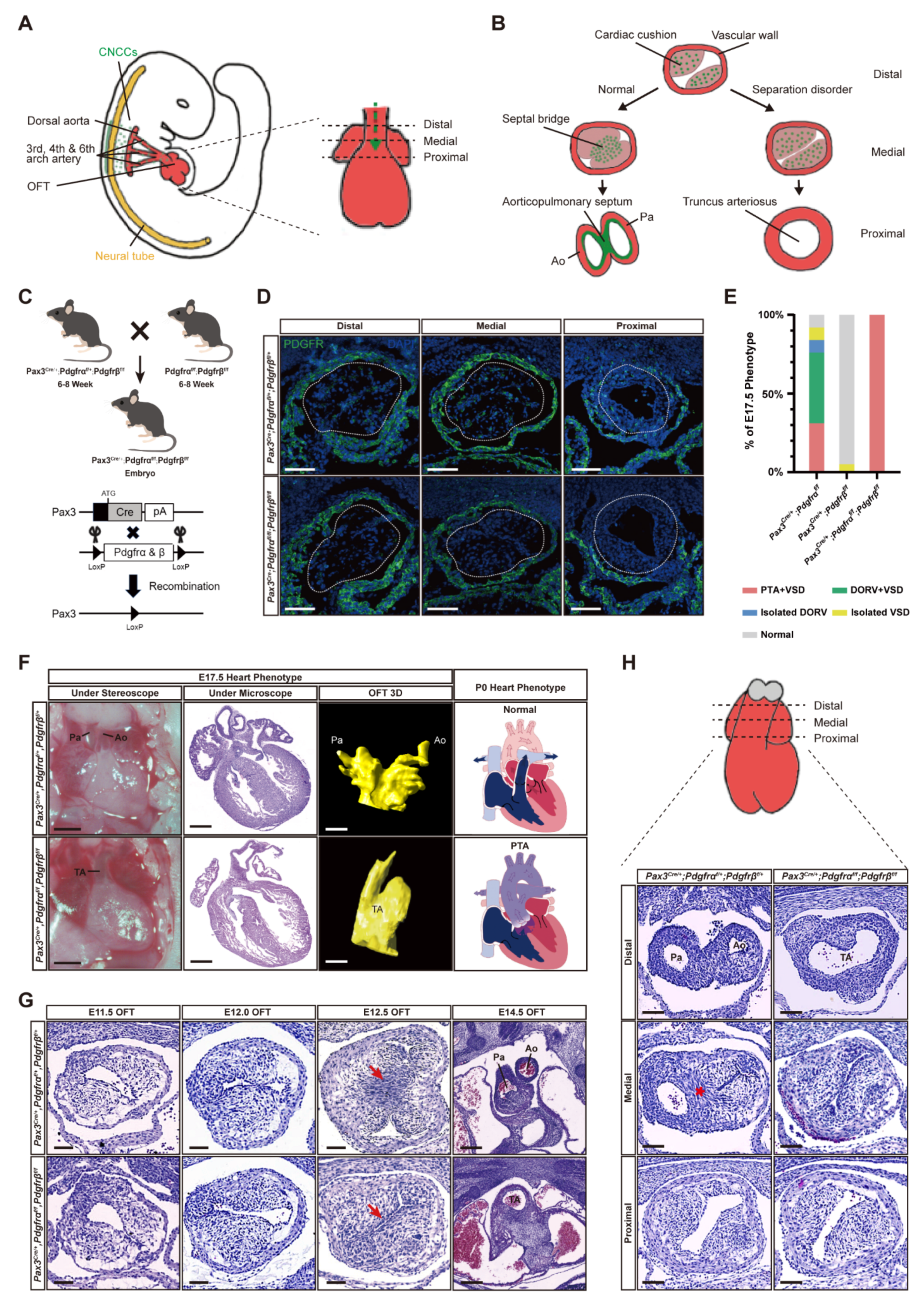
3.1. Simultaneous Knockout of PDGFRα and PDGFRβ in Pax3+ NCCs Induced PTA
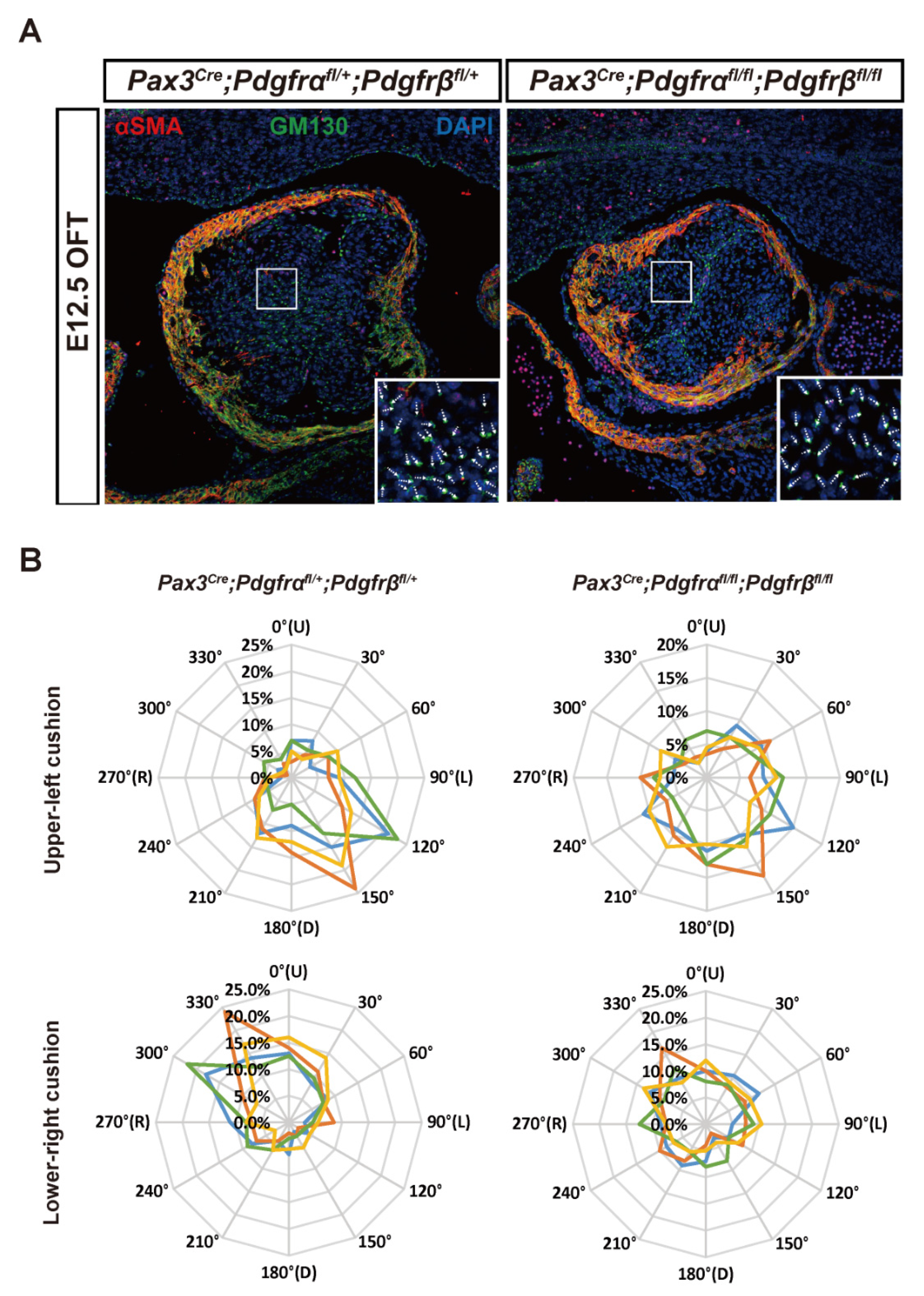
3.2. Condensation Failure of CNCC-Derived Cells Due to Disturbance of Cell Polarity after Pdgfrα and Pdgfrβ Knockout
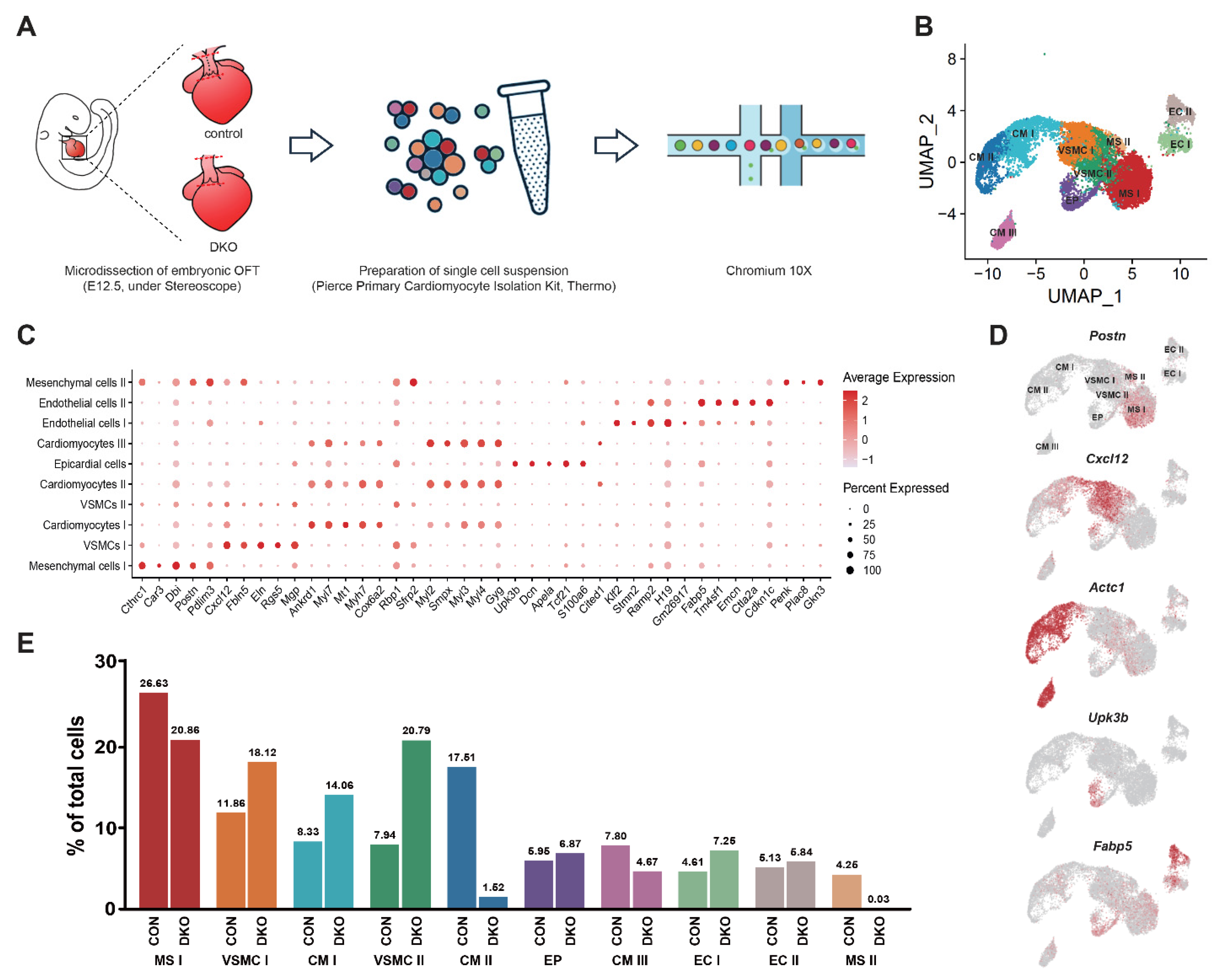
3.3. Single-Cell Transcriptomic Sequencing and Overview of Developing OFT Cells
3.4. The Disturbance of CMs Trans-Differentiation after Pdgfrα and Pdgfrβ Knockout
3.5. Absence of a Key MS Cluster Associated with Septal Bridge Formation after Pdgfrα and Pdgfrβ Knockout
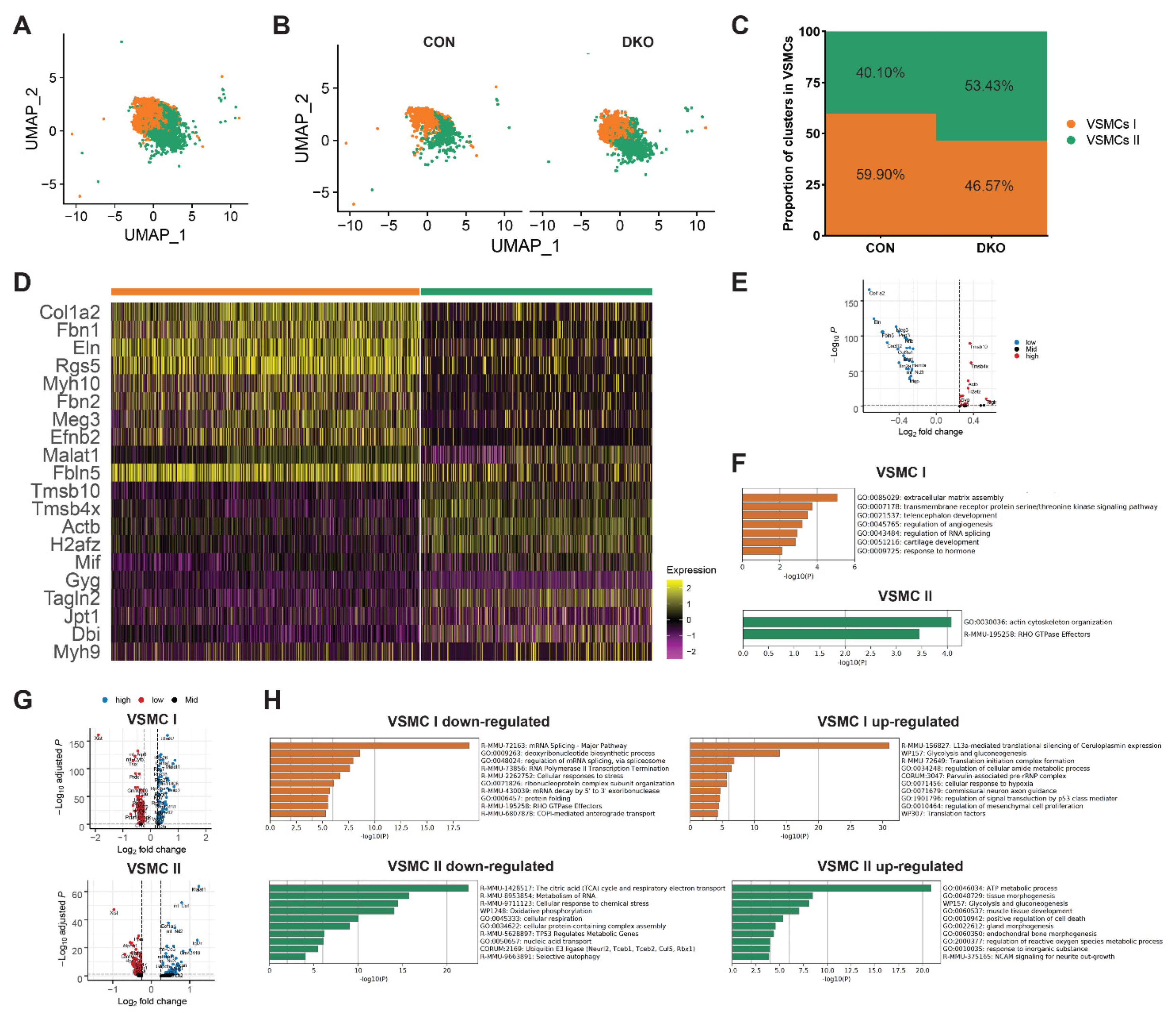
3.6. More VSMC in an Immature State after PDGFRα and PDGFRβ Knockout
3.7. Ligand-Receptor Interaction Changes among MS, CM and VSMC Clusters after PDGFRα and PDGFRβ Knockout
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| PTA | persistent truncus arteriosus |
| CHD | congenital heart disease |
| OFT | outflow tract |
| CNCC | cardiac neural crest cell |
| DKO | double knockout |
| VSMC | vascular smooth muscle cell |
| CM | cardiomyocyte |
| MS | mesenchymal cell |
References
- Srivastava, D. Making or Breaking the Heart: From Lineage Determination to Morphogenesis. Cell 2006, 126, 1037–1048. [Google Scholar] [CrossRef]
- Slavik, Z.; Keeton, B.R.; Salmon, A.P.; Sutherland, G.R.; Fong, L.V.; Monro, J.L. Persistent truncus arteriosus operated during infancy: Long-term follow-up. Pediatr. Cardiol. 1994, 15, 112–115. [Google Scholar] [CrossRef]
- Pearl, J.M.; Laks, H.; Drinkwater, D.C.; Milgalter, E.; Charas, O.A.; Giacobetti, F.; George, B.; Williams, R. Repair of truncus arteriosus in infancy. Ann. Thorac. Surg. 1991, 52, 780–786. [Google Scholar] [CrossRef]
- Nichols, D.G.; Ungerleider, R.M.; Spevak, P.J. Critical Heart Disease in Infants and Children (Second Edition); Mosby: Philadelphia, PA, USA, 2006; pp. 995–1024. [Google Scholar]
- Rajasinghe, H.A.; McElhinney, D.B.; Reddy, V.; Mora, B.N.; Hanley, F.L. Long-term follow-up of truncus arteriosus repaired in infancy: A twenty-year experience. J. Thorac. Cardiovasc. Surg. 1997, 113, 869–879. [Google Scholar] [CrossRef]
- Etchevers, H.C.; Vincent, C.; Le Douarin, N.M.; Couly, G.F. The cephalic neural crest provides pericytes and smooth muscle cells to all blood vessels of the face and forebrain. Development 2001, 128, 1059–1068. [Google Scholar] [CrossRef]
- Bhatt, S.; Diaz, R.; Trainor, P. Signals and Switches in Mammalian Neural Crest Cell Differentiation. Cold Spring Harb. Perspect. Biol. 2013, 5, a008326. [Google Scholar] [CrossRef]
- Szabó, A.; Mayor, R. Mechanisms of Neural Crest Migration. Annu. Rev. Genet. 2018, 52, 43–63. [Google Scholar] [CrossRef]
- Plein, A.; Fantin, A.; Ruhrberg, C. Neural Crest Cells in Cardiovascular Development. Curr. Top. Dev. Biol. 2015, 111, 183–200. [Google Scholar] [CrossRef]
- Kirby, M.L.; Gale, T.F.; Stewart, D.E. Neural Crest Cells Contribute to Normal Aorticopulmonary Septation. Science 1983, 220, 1059–1061. [Google Scholar] [CrossRef]
- Le Lièvre, C.; Le Douarin, N. Mesenchymal derivatives of the neural crest: Analysis of chimaeric quail and chick embryos. J. Embryol. Exp. Morphol. 1975, 34, 125–154. [Google Scholar] [CrossRef]
- Brickner, M.E.; Hillis, L.D.; Lange, R.A. Congenital heart disease in adults. First of two parts. N. Engl. J. Med. 2000, 342, 256–263. [Google Scholar] [CrossRef]
- Darrigrand, J.F.; Valente, M.; Comai, G.; Martinez, P.; Petit, M.; Nishinakamura, R.; Osorio, D.S.; Renault, G.; Marchiol, C.; Ribes, V.; et al. Dullard-mediated Smad1/5/8 inhibition controls mouse cardiac neural crest cells condensation and outflow tract septation. eLife 2020, 9, e50325. [Google Scholar] [CrossRef]
- Hamblet, N.; Lijam, N.; Ruiz-Lozano, P. Dishevelled 2 is essential for cardiac outflo w tract development, somite segmentation and neural tube closure. Development 2002, 129, 5827–5838. [Google Scholar] [CrossRef]
- Sauka-Spengler, T.; Bronner-Fraser, M. A gene regulatory network orchestrates neural crest formation. Nat. Rev. Mol. Cell Biol. 2008, 9, 557–568. [Google Scholar] [CrossRef]
- Van Roy, F.; Maruhashi, M.; Francis, A.; Nelles, L.; Kondoh, H.; Huylebroeck, K.; Higashi, Y. Faculty Opinions recommendation of Mice lacking ZFHX1B, the gene that codes for Smad-interacting protein-1, reveal a role for multiple neural crest cell defects in the etiology of Hirschsprung disease-mental retardation syndrome. Am. J. Hum. Genet. 2003, 72, 465–470. [Google Scholar] [CrossRef]
- Feiner, L.; Webber, A.L.; Brown, C.B.; Lu, M.M.; Jia, L.; Feinstein, P.; Mombaerts, P.; Epstein, J.A.; Raper, J.A. Targeted disruption of semaphorin 3C leads to persistent truncus arteriosus and aortic arch interruption. Development 2001, 128, 3061–3070. [Google Scholar] [CrossRef]
- Toyofuku, T.; Yoshida, J.; Sugimoto, T.; Yamamoto, M.; Makino, N.; Takamatsu, H.; Takegahara, N.; Suto, F.; Hori, M.; Fujisawa, H.; et al. Repulsive and attractive semaphorins cooperate to direct the navigation of cardiac neural crest cells. Dev. Biol. 2008, 321, 251–262. [Google Scholar] [CrossRef]
- Hollinger, J.O.; Hart, C.E.; Hirsch, S.N.; Lynch, S.; Friedlaender, G.E. Recombinant Human Platelet-Derived Growth Factor: Biology and Clinical Applications. J. Bone Jt. Surg. 2008, 90, 48–54. [Google Scholar] [CrossRef]
- Andrae, J.; Gallini, R.; Betsholtz, C. Role of platelet-derived growth factors in physiology and medicine. Genes Dev. 2008, 22, 1276–1312. [Google Scholar] [CrossRef]
- Richarte, A.M.; Mead, H.B.; Tallquist, M.D. Cooperation between the PDGF receptors in cardiac neural crest cell migration. Dev. Biol. 2007, 306, 785–796. [Google Scholar] [CrossRef] [Green Version]
- Council, N.R. Guide for the Care and Use of Laboratory Animals; The National Academies Press: Washington, DC, USA, 1996. [Google Scholar]
- Butler, A.; Hoffman, P.; Smibert, P.; Papalexi, E.; Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018, 36, 411–420. [Google Scholar] [CrossRef]
- Becht, E.; McInnes, L.; Healy, J.; Dutertre, C.-A.; Kwok, I.W.H.; Ng, L.G.; Ginhoux, F.; Newell, E.W. Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol. 2018, 37, 38–44. [Google Scholar] [CrossRef]
- Bergen, V.; Lange, M.; Peidli, S.; Wolf, F.A.; Theis, F.J. Generalizing RNA velocity to transient cell states through dynamical modeling. Nat. Biotechnol. 2020, 38, 1408–1414. [Google Scholar] [CrossRef]
- Jin, S.; Guerrero-Juarez, C.F.; Zhang, L.; Chang, I.; Ramos, R.; Kuan, C.-H.; Myung, P.; Plikus, M.V.; Nie, Q. Inference and analysis of cell-cell communication using CellChat. Nat. Commun. 2021, 12, 1–20. [Google Scholar] [CrossRef]
- Farbehi, N.; Patrick, R.; Dorison, A.; Xaymardan, M.; Janbandhu, V.; Wystub-Lis, K.; Ho, J.W.; E Nordon, R.; Harvey, R.P. Single-cell expression profiling reveals dynamic flux of cardiac stromal, vascular and immune cells in health and injury. eLife 2019, 8, e43882. [Google Scholar] [CrossRef]
- Muzumdar, M.D.; Tasic, B.; Miyamichi, K.; Li, L.; Luo, L. A global double-fluorescent Cre reporter mouse. Genesis 2007, 45, 593–605. [Google Scholar] [CrossRef]
- Chen, W.; Liu, X.; Li, W.; Shen, H.; Zeng, Z.; Yin, K.; Priest, J.R.; Zhou, Z. Single-cell transcriptomic landscape of cardiac neural crest cell derivatives during development. EMBO Rep. 2021, 22, e52389. [Google Scholar] [CrossRef]
- Rudat, C.; Grieskamp, T.; Röhr, C.; Airik, R.; Wrede, C.; Hegermann, J.; Herrmann, B.G.; Schuster-Gossler, K.; Kispert, A. Upk3b Is Dispensable for Development and Integrity of Urothelium and Mesothelium. PLoS ONE 2014, 9, e112112. [Google Scholar] [CrossRef]
- Teruhide, K.; Takayuki, S.; Akiko, K.; Ichikawa-Shindo, Y.; Kawate, H.; Shindo, T. Adrenomedullin-RAMP2 System in Vascular Endothelial Cells. J. Atheroscler. Thromb. 2015, 22, 647–653. [Google Scholar]
- Li, G.; Xu, A.; Sim, S.; Priest, J.R.; Tian, X.; Khan, T.; Quertermous, T.; Zhou, B.; Tsao, P.S.; Quake, S.R.; et al. Transcriptomic Profiling Maps Anatomically Patterned Subpopulations among Single Embryonic Cardiac Cells. Dev. Cell 2016, 39, 491–507. [Google Scholar] [CrossRef]
- Wilczewski, C.M.; Hepperla, A.J.; Shimbo, T.; Wasson, L.; Robbe, Z.L.; Davis, I.J.; Wade, P.A.; Conlon, F.L. CHD4 and the NuRD complex directly control cardiac sarcomere formation. Proc. Natl. Acad. Sci. USA 2018, 115, 6727–6732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivins, S.; Chappell, J.; Vernay, B.; Suntharalingham, J.; Martineau, A.; Mohun, T.J.; Scambler, P.J. The CXCL12/CXCR4 Axis Plays a Critical Role in Coronary Artery Development. Dev. Cell 2015, 33, 455–468. [Google Scholar] [CrossRef] [PubMed]
- Daniel, J.-M.; Prock, A.; Dutzmann, J.; Sonnenschein, K.; Thum, T.; Bauersachs, J.; Sedding, D.G. Regulator of G-Protein Signaling 5 Prevents Smooth Muscle Cell Proliferation and Attenuates Neointima Formation. Arter. Thromb. Vasc. Biol. 2016, 36, 317–327. [Google Scholar] [CrossRef]
- Gunaje, J.J.; Bahrami, A.J.; Schwartz, S.M.; Daum, G.; Jr, W.M.M. PDGF-dependent regulation of regulator of G protein signaling-5 expression and vascular smooth muscle cell functionality. Am. J. Physiol. Physiol. 2011, 301, C478–C489. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Chen, W.; Li, W.; Li, Y.; Priest, J.R.; Zhou, B.; Wang, J.; Zhou, Z. Single-Cell RNA-Seq of the Developing Cardiac Outflow Tract Reveals Convergent Development of the Vascular Smooth Muscle Cells. Cell Rep. 2019, 28, 1346–1361.e4. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, N.; Roux, M.; Ryckebüsch, L.; Niederreither, K.; Dollé, P.; Moon, A.; Capecchi, M.; Zaffran, S. Hox genes define distinct progenitor sub-domains within the second heart field. Dev. Biol. 2011, 353, 266–274. [Google Scholar] [CrossRef] [PubMed]
- Plein, A.; Calmont, A.; Fantin, A.; Denti, L.; Anderson, N.A.; Scambler, P.J.; Ruhrberg, C. Neural crest–derived SEMA3C activates endothelial NRP1 for cardiac outflow tract septation. J. Clin. Investig. 2015, 125, 2661–2676. [Google Scholar] [CrossRef]
- Du, Y.; Zhang, Z.; Xiong, W.; Li, N.; Liu, H.; He, H.; Li, Q.; Liu, Y.; Zhang, L. Estradiol promotes EMT in endometriosis via MALAT1/miR200s sponge function. Reproduction 2019, 157, 179–188. [Google Scholar] [CrossRef]
- Kestens, C.; Siersema, P.D.; Offerhaus, G.J.A.; Van Baal, J.W.P.M. BMP4 Signaling Is Able to Induce an Epithelial-Mesenchymal Transition-Like Phenotype in Barrett’s Esophagus and Esophageal Adenocarcinoma through Induction of SNAIL2. PLoS ONE 2016, 11, e0155754. [Google Scholar] [CrossRef]
- Lin, Y.; Ma, Q.; Li, L.; Wang, H. The CXCL12-CXCR4 axis promotes migration, invasiveness, and EMT in human papillary thyroid carcinoma B-CPAP cells via NF-κB signaling. Biochemistry and cell biology = Biochimie et biologie cellulaire. Biochem. Cell Biol. 2018, 96, 619–626. [Google Scholar] [CrossRef]
- Pang, L.; Li, Q.; Li, S.; He, J.; Cao, W.; Lan, J.; Sun, B.; Zou, H.; Wang, C.; Liu, R.; et al. Membrane type 1-matrix metalloproteinase induces epithelial-to-mesenchymal transition in esophageal squamous cell carcinoma: Observations from clinical and in vitro analyses. Sci. Rep. 2016, 6, 22179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiota, M.; Zardan, A.; Takeuchi, A.; Kumano, M.; Beraldi, E.; Naito, S.; Zoubeidi, A.; Gleave, M.E. Clusterin Mediates TGF-β–Induced Epithelial–Mesenchymal Transition and Metastasis via Twist1 in Prostate Cancer Cells. Cancer Res. 2012, 72, 5261–5272. [Google Scholar] [CrossRef] [PubMed]
- Yin, W.; Zhu, H.; Tan, J.; Xin, Z.; Zhou, Q.; Cao, Y.; Wu, Z.; Wang, L.; Zhao, M.; Jiang, X.; et al. Identification of collagen genes related to immune infiltration and epithelial-mesenchymal transition in glioma. Cancer Cell Int. 2021, 21, 276. [Google Scholar] [CrossRef]
- Nicolas, C.; Brian, L.; Syandan, C.; Malathi, C.; Nenad, B.; Leong, K.W. Induced pluripotent stem cell-derived cardiac progenitors differentiate to cardiomyocytes and form biosynthetic tissues. PLoS ONE 2013, 8, e65963. [Google Scholar]
- Jain, R.; Li, D.; Gupta, M.; Manderfield, L.J.; Ifkovits, J.L.; Wang, Q.; Liu, F.; Liu, Y.; Poleshko, A.; Padmanabhan, A.; et al. Integration of Bmp and Wnt signaling by Hopx specifies commitment of cardiomyoblasts. Science 2015, 348, aaa6071. [Google Scholar] [CrossRef]
- La Manno, G.; Soldatov, R.; Zeisel, A.; Braun, E.; Hochgerner, H.; Petukhov, V.; Lidschreiber, K.; Kastriti, M.E.; Lönnerberg, P.; Furlan, A.; et al. RNA velocity of single cells. Nature 2018, 560, 494–498. [Google Scholar] [CrossRef]
- Vincentz, J.W.; Firulli, B.A.; Lin, A.; Spicer, D.B.; Howard, M.J.; Firulli, A.B. Twist1 Controls a Cell-Specification Switch Governing Cell Fate Decisions within the Cardiac Neural Crest. PLoS Genet. 2013, 9, e1003405. [Google Scholar] [CrossRef]
- Vincentz, J.W.; Barnes, R.M.; Rodgers, R.; Firulli, B.A.; Conway, S.J.; Firulli, A.B. An absence of Twist1 results in aberrant cardiac neural crest morphogenesis. Dev. Biol. 2008, 320, 131–139. [Google Scholar] [CrossRef]
- Gitler, A.D.; Lu, M.M.; A Epstein, J. PlexinD1 and Semaphorin Signaling Are Required in Endothelial Cells for Cardiovascular Development. Dev. Cell 2004, 7, 107–116. [Google Scholar] [CrossRef]
- Civitarese, R.A.; Kapus, A.; McCulloch, C.A.; Connelly, K.A. Role of integrins in mediating cardiac fibroblast–cardiomyocyte cross talk: A dynamic relationship in cardiac biology and pathophysiology. Basic Res. Cardiol. 2016, 112, 6. [Google Scholar] [CrossRef]
- Tirosh-Finkel, L.; Zeisel, A.; Brodt-Ivenshitz, M.; Shamai, A.; Yao, Z.; Seger, R.; Domany, E.; Tzahor, E. BMP-mediated inhibition of FGF signaling promotes cardiomyocyte differentiation of anterior heart field progenitors. Development 2010, 137, 2989–3000. [Google Scholar] [CrossRef] [PubMed]
- D’Amato, G.; Luxán, G.; de la Pompa, J.L. Notch signalling in ventricular chamber development and cardiomyopathy. FEBS J. 2016, 283, 4223–4237. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, D.; Van Berlo, J. The Role of TGF-β Signaling in Cardiomyocyte Proliferation. Curr. Heart Fail. Rep. 2020, 17, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Bassat, E.; Mutlak, Y.E.; Genzelinakh, A.; Shadrin, I.; Umansky, K.B.; Yifa, O.; Kain, D.; Rajchman, D.; Leach, J.; Bassat, D.R.; et al. The extracellular matrix protein agrin promotes heart regeneration in mice. Nature 2017, 547, 179–184. [Google Scholar] [CrossRef]
- Lauten, A.; Gerhard-Garcia, A.; Suhr, F.; Fischer, J.H.; Figulla, H.R.; Bloch, W. Impact of Ischemia-Reperfusion on Extracellular Matrix Processing and Structure of the Basement Membrane of the Heart. PLoS ONE 2014, 9, e92833. [Google Scholar] [CrossRef]
- Costell, M.; Carmona, R.; Gustafsson, E.; González-Iriarte, M.; Fässler, R.; Muñoz-Chápuli, R. Hyperplastic conotruncal endocardial cushions and transposition of great arteries in perlecan-null mice. Circ. Res. 2002, 91, 158–164. [Google Scholar] [CrossRef]
- Yu, S.; Crawford, D.; Tsuchihashi, T.; Behrens, T.W.; Srivastava, D. The chemokine receptor CXCR7 functions to regulate cardiac valve remodeling. Dev. Dyn. 2011, 240, 384–393. [Google Scholar] [CrossRef]
- Sierro, F.; Biben, C.; Martínez-Muñoz, L.; Mellado, M.; Ransohoff, R.M.; Li, M.; Woehl, B.; Leung, H.; Groom, J.; Batten, M.; et al. Disrupted cardiac development but normal hematopoiesis in mice deficient in the second CXCL12/SDF-1 receptor, CXCR7. Proc. Natl. Acad. Sci. USA 2007, 104, 14759–14764. [Google Scholar] [CrossRef]
- Chen, E.; Hermanson, S.; Ekker, S.C. Syndecan-2 is essential for angiogenic sprouting during zebrafish development. Blood 2004, 103, 1710–1719. [Google Scholar] [CrossRef]
- Chen, L.; Klass, C.; Woods, A. Syndecan-2 regulates transforming growth factor-beta signaling. J. Biol. Chem. 2004, 279, 15715–15718. [Google Scholar] [CrossRef]
- Noguer, O.; Villena, J.; Lorita, J.; Vilaró, S.; Reina, M. Syndecan-2 downregulation impairs angiogenesis in human microvascular endothelial cells. Exp. Cell Res. 2009, 315, 795–808. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.-H.I.; Feinstein, T.N.; Jha, A.; McCleary, J.T.; Xu, J.; Arrigo, A.B.; Rong, G.; MacLay, L.M.; Ridge, T.; Xu, X.; et al. Mutation of LRP1 in cardiac neural crest cells causes congenital heart defects by perturbing outflow lengthening. Commun. Biol. 2020, 3, 312. [Google Scholar] [CrossRef] [PubMed]
- Bajolle, F.; Zaffran, S.; Meilhac, S. Myocardium at the base of the aorta and pulmonary trunk is prefi gured in the outflow tract of the heart and in subdomains of the second heart field. Dev. Biol. 2008, 313, 25–34. [Google Scholar] [CrossRef]
- Waldo, K.; Miyagawa-Tomita, S.; Kumiski, D. Cardiac neural crest cells provide new insight into septation of the cardiac outflow tract: Aortic sac to ventricular septal closure. Dev. Biol. 1998, 196, 129–144. [Google Scholar] [CrossRef] [PubMed]
- Morrison-Graham, K.; Schatteman, G.C.; Bork, T.; Bowen-Pope, D.F.; A Weston, J. A PDGF receptor mutation in the mouse (Patch) perturbs the development of a non-neuronal subset of neural crest-derived cells. Development 1992, 115, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Tallquist, M.D.; Soriano, P. Cell autonomous requirement for PDGFRalpha in populations of cranial and cardiac neural crest cells. Development 2003, 130, 507–518. [Google Scholar] [CrossRef] [PubMed]
- Soriano, P. Abnormal kidney development and hematological disorders in PDGF beta-receptor mutant mice. Genes Dev. 1994, 8, 1888–1896. [Google Scholar] [CrossRef]
- de Soysa, T.; Ranade, S.S.; Okawa, S.; Ravichandran, S.; Huang, Y.; Salunga, H.T.; Schricker, A.; del Sol, A.; Gifford, C.A.; Srivastava, D. Single-cell analysis of cardiogenesis reveals basis for organ-level developmental defects. Nature 2019, 572, 120–124. [Google Scholar] [CrossRef]
- Ya, J.; Hoff, M.J.B.V.D.; de Boer, P.A.J.; Tesink-Taekema, S.; Franco, D.; Moorman, A.F.M.; Lamers, W.H. Normal Development of the Outflow Tract in the Rat. Circ. Res. 1998, 82, 464–472. [Google Scholar] [CrossRef]
- Anderson, R.H.; Webb, S.; Brown, N.A.; Lamers, W.; Moorman, A. Development of the heart: (3) Formation of the ventricular outflow tracts, arterial valves, and intrapericardial arterial trunks. Heart 2003, 89, 1110–1118. [Google Scholar] [CrossRef]
- Armstrong, J.J.; Larina, I.V.; Dickinson, M.E.; Zimmer, W.E.; Hirschi, K.K. Characterization of bacterial artificial chromosome transgenic mice expressing mCherry fluorescent protein substituted for the murine smooth muscle alpha-actin gene. Genesis 2010, 48, 457–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, T.; Song, S.; Jiang, H.; Lian, H.; Hu, S. Single Cell Sequencing Reveals Mechanisms of Persistent Truncus Arteriosus Formation after PDGFRα and PDGFRβ Double Knockout in Cardiac Neural Crest Cells. Genes 2022, 13, 1708. https://doi.org/10.3390/genes13101708
Chen T, Song S, Jiang H, Lian H, Hu S. Single Cell Sequencing Reveals Mechanisms of Persistent Truncus Arteriosus Formation after PDGFRα and PDGFRβ Double Knockout in Cardiac Neural Crest Cells. Genes. 2022; 13(10):1708. https://doi.org/10.3390/genes13101708
Chicago/Turabian StyleChen, Tianyun, Shen Song, Haobin Jiang, Hong Lian, and Shengshou Hu. 2022. "Single Cell Sequencing Reveals Mechanisms of Persistent Truncus Arteriosus Formation after PDGFRα and PDGFRβ Double Knockout in Cardiac Neural Crest Cells" Genes 13, no. 10: 1708. https://doi.org/10.3390/genes13101708
APA StyleChen, T., Song, S., Jiang, H., Lian, H., & Hu, S. (2022). Single Cell Sequencing Reveals Mechanisms of Persistent Truncus Arteriosus Formation after PDGFRα and PDGFRβ Double Knockout in Cardiac Neural Crest Cells. Genes, 13(10), 1708. https://doi.org/10.3390/genes13101708