
Potential CRISPR Base Editing Therapeutic Options in a Sorsby Fundus Dystrophy Patient

Abstract
:1. Introduction
2. Materials and Methods
2.1. Clinical Examination
2.2. Genetic Testing
2.3. Base Editing
3. Results
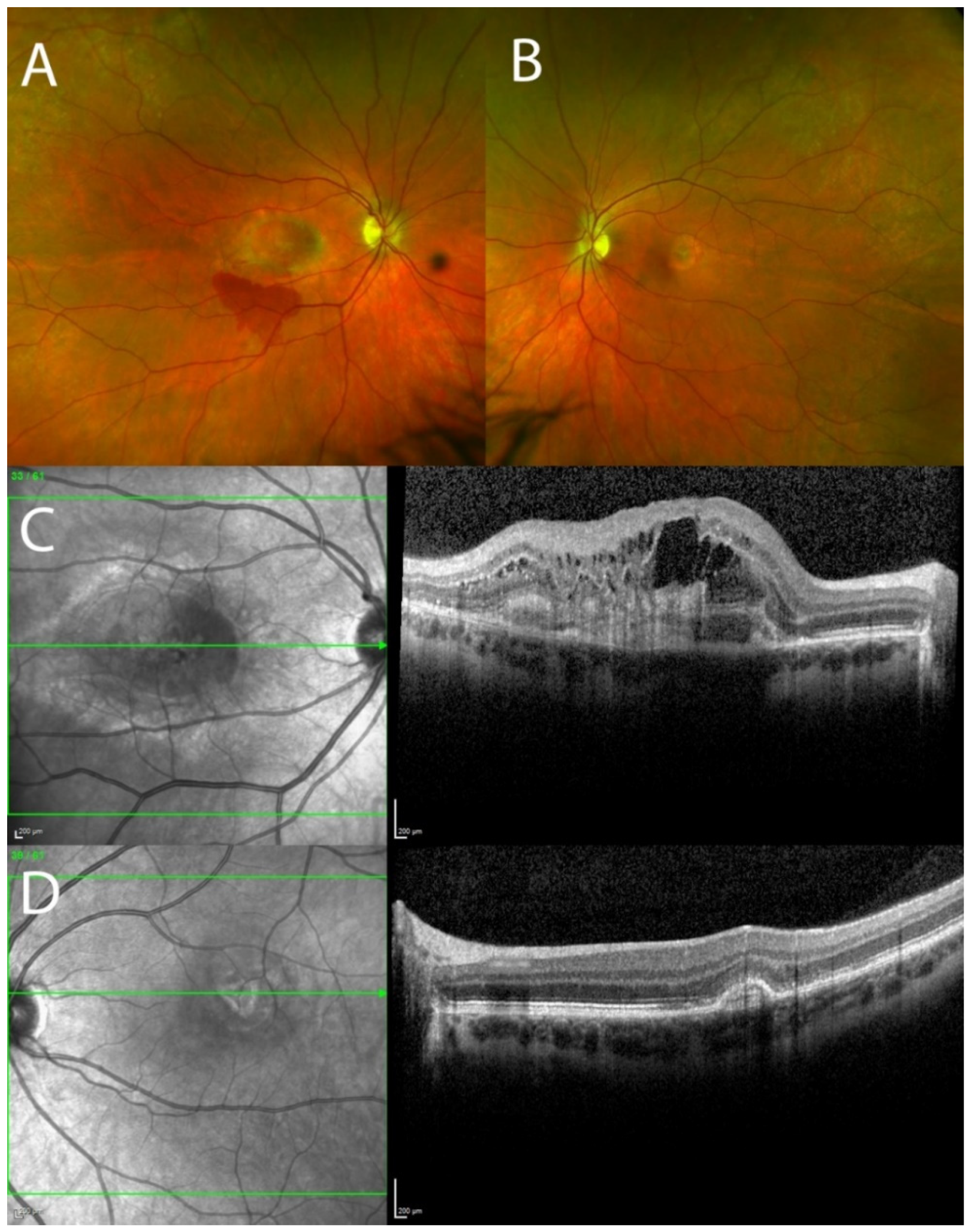
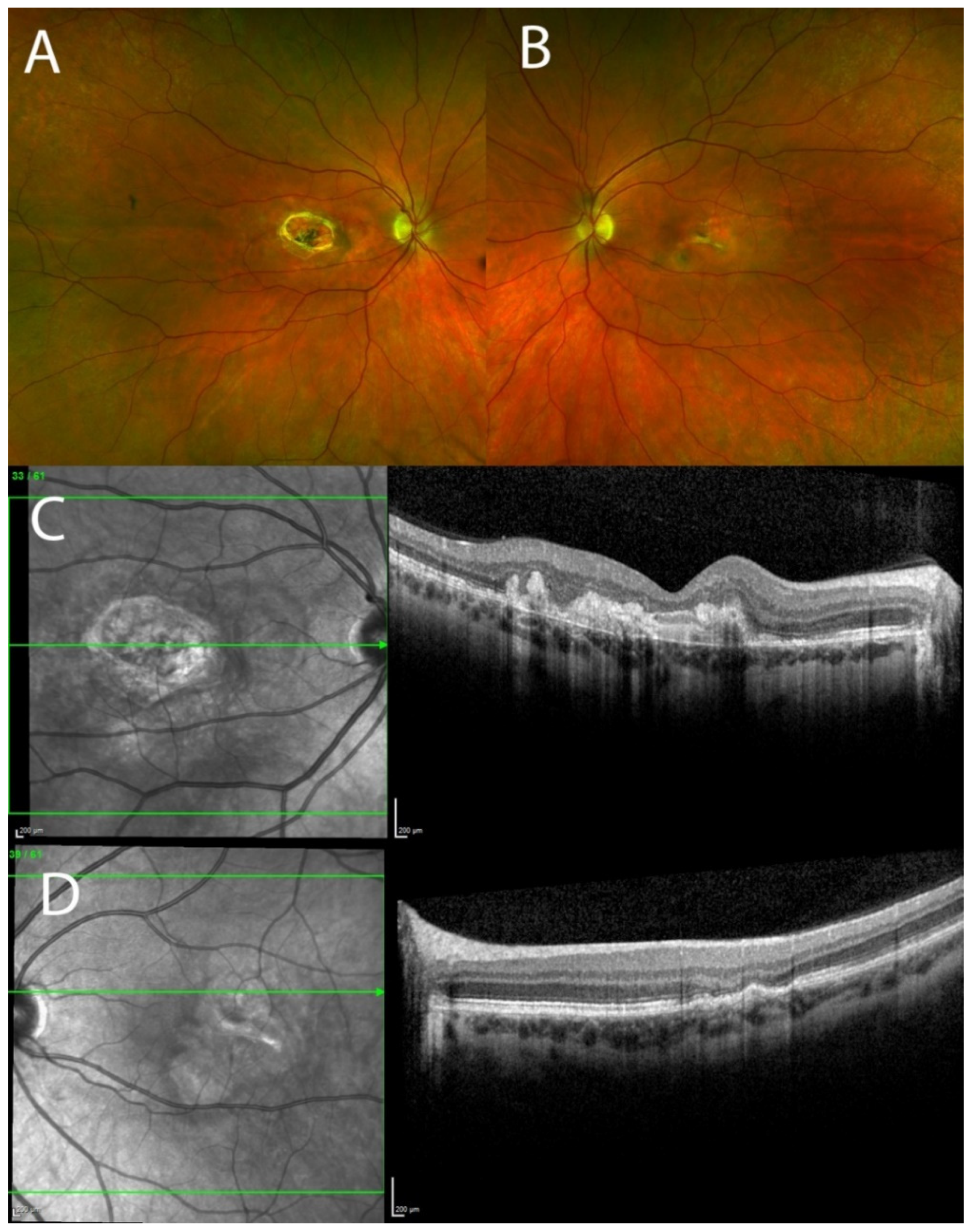
3.1. Clinical Results
3.2. Genetic Testing
3.3. Base Editing
4. Discussion
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dewing, J.M.; Carare, R.O.; Lotery, A.J.; Ratnayaka, J.A. The Diverse Roles of TIMP-3: Insights into Degenerative Diseases of the Senescent Retina and Brain. Cells 2019, 9, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anand-Apte, B.; Chao, J.R.; Singh, R.; Stohr, H. Sorsby fundus dystrophy: Insights from the past and looking to the future. J. Neurosci. Res. 2019, 97, 88–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gliem, M.; Muller, P.L.; Mangold, E.; Holz, F.G.; Bolz, H.J.; Stöhr, H.; Weber, B.H.F. Sorsby Fundus Dystrophy: Novel Mutations, Novel Phenotypic Characteristics, and Treatment Outcomes. Investig. Ophthalmol. Vis. Sci. 2015, 56, 2664–2676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langton, K.P.; McKie, N.; Smith, B.M.; Brown, N.J.; Barker, M.D. Sorsby’s fundus dystrophy mutations impair turnover of TIMP-3 by retinal pigment epithelial cells. Hum. Mol. Genet. 2005, 14, 3579–3586. [Google Scholar] [CrossRef] [Green Version]
- Apte, S.S.; Mattei, M.G.; Olsen, B.R. Cloning of the cDNA encoding human tissue inhibitor of metalloproteinases-3 (TIMP-3) and mapping of the TIMP3 gene to chromosome 22. Genomics 1994, 19, 86–90. [Google Scholar] [CrossRef]
- Qi, J.H.; Ebrahem, Q.; Moore, N.; Murphy, G.; Claesson-Welsh, L.; Bond, M.; Baker, A.; Anand-Apte, B. A novel function for tissue inhibitor of metalloproteinases-3 (TIMP3): Inhibition of angiogenesis by blockage of VEGF binding to VEGF receptor-2. Nat. Med. 2003, 9, 407–415. [Google Scholar] [CrossRef]
- Fogarasi, M.; Janssen, A.; Weber, B.H.; Stohr, H. Molecular dissection of TIMP3 mutation S156C associated with Sorsby fundus dystrophy. Matrix Biol. 2008, 27, 381–392. [Google Scholar] [CrossRef]
- Qi, J.H.; Dai, G.; Luthert, P.; Chaurasia, S.; Hollyfield, J.; Weber, B.H.; Stöhr, H.; Anand-Apte, B. S156C mutation in tissue inhibitor of metalloproteinases-3 induces increased angiogenesis. J. Biol. Chem. 2009, 284, 19927–19936. [Google Scholar] [CrossRef] [Green Version]
- Russell, S.; Bennett, J.; Wellman, J.A.; Chung, D.C.; Yu, Z.F.; Tillman, A.; Wittes, J.; Pappas, J.; Elci, O.; McCague, S.; et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: A randomised, controlled, open-label, phase 3 trial. Lancet 2017, 390, 849–860. [Google Scholar] [CrossRef]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable base editing of A*T to G*C in genomic DNA without DNA cleavage. Nature 2017, 551, 464–471. [Google Scholar] [CrossRef]
- Kurt, I.C.; Zhou, R.; Iyer, S.; Garcia, S.P.; Milletr, B.R.; Langner, L.M.; Grünewald, J.; Joung, J.K. CRISPR C-to-G base editors for inducing targeted DNA transversions in human cells. Nat. Biotechnol. 2021, 39, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyaoka, Y.; Berman, J.R.; Cooper, S.B.; Mayerl, S.J.; Chan, A.H.; Zhang, B.; Karlin-Neumann, G.A.; Conklin, B.R. Systematic quantification of HDR and NHEJ reveals effects of locus, nuclease, and cell type on genome-editing. Sci. Rep. 2016, 6, 23549. [Google Scholar] [CrossRef] [Green Version]
- Richter, M.F.; Zhao, K.T.; Eton, E.; Lapinaite, A.; Newby, G.A.; Thuronyi, B.W.; Wilson, C.; Koblan, L.; Zeng, J.; Bauer, D.E.; et al. Phage-assisted evolution of an adenine base editor with improved Cas domain compatibility and activity. Nat. Biotechnol. 2020, 38, 883–891. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Chemparathy, A.; Zeng, L.; Kempton, H.R.; Shang, S.; Nakamura, M.; Qi, L.S. Engineered miniature CRISPR-Cas system for mammalian genome regulation and editing. Mol. Cell 2021, 81, 4333–4345. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.H.; Miller, S.M.; Geurts, M.H.; Tang, W.; Chen, L.; Sun, N.; Zeina, C.M.; Gao, X.; Rees, H.A.; Lin, Z.; et al. Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature 2018, 556, 57–63. [Google Scholar] [CrossRef]
- Komor, A.C.; Zhao, K.T.; Packer, M.S.; Gaudelli, N.M.; Waterbury, A.L.; Koblan, L.W.; Kim, Y.B.; Badran, A.H.; Liu, D.R. Improved base excision repair inhibition and bacteriophage Mu Gam protein yields C:G-to-T:A base editors with higher efficiency and product purity. Sci. Adv. 2017, 3, eaao4774. [Google Scholar] [CrossRef] [Green Version]
- Sun, N.; Zhao, D.; Li, S.; Et Zhang, Z.; Bi, C.; Zhalng, X. Reconstructed glycosylase base editors GBE2.0 with enhanced C-to-G base editing efficiency and purity. Mol. Ther. 2022, 30, 2452–2463. [Google Scholar] [CrossRef]
- Ng, P.C.; Henikoff, S. Predicting deleterious amino acid substitutions. Genome Res. 2001, 11, 863–874. [Google Scholar] [CrossRef] [Green Version]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Et Ramensky, V.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef] [PubMed]
- Christensen, D.R.G.; Brown, F.E.; Cree, A.J.; Ratnayaka, J.A.; Lotetry, A.J. Sorsby fundus dystrophy—A review of pathology and disease mechanisms. Exp. Eye Res. 2017, 165, 35–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wijesuriya, S.D.; Evans, K.; Jay, M.R.; Davison, C.; Wetber, B.H.; Bird, A.C.; Bhattacharya, S.S.; Gregory, C.Y. Sorsby’s fundus dystrophy in the British Isles: Demonstration of a striking founder effect by microsatellite-generated haplotypes. Genome Res. 1996, 6, 92–101. [Google Scholar] [CrossRef] [Green Version]
- Menassa, N.; Burgula, S.; Empeslidis, T.; Tsaousis, K.T. Bilateral choroidal neovascular membrane in a young patient with Sorsby fundus dystrophy: The value of prompt treatment. BMJ Case Rep. 2017, 2017, bcr-2017. [Google Scholar] [CrossRef] [PubMed]
- Gemenetzi, M.K.; Luff, A.J.; Lotery, A.J. Successful treatment of choroidal neovascularization secondary to sorsby fundus dystrophy with intravitreal bevacizumab. Retin. Cases Brief Rep. 2011, 5, 132–135. [Google Scholar] [CrossRef]
- Keller, J.; Giralt, J.; Alforja, S.; Casaroli-Marano, R.P. Altering the clinical course of Sorsby fundus dystrophy with the use of anti-vascular endothelial growth factor intraocular therapy. Retin. Cases Brief Rep. 2015, 9, 104–105. [Google Scholar] [CrossRef]
- Warwick, A.; Gibson, J.; Sood, R.; Lotery, A. A rare penetrant TIMP3 mutation confers relatively late onset choroidal neovascularisation which can mimic age-related macular degeneration. Eye 2016, 30, 488–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, D.H.; Talaga, K.C.; Rivest, A.J.; Barron, E.; Hageman, G.S.; Johnson, L.V. Characterization of β amyloid assemblies in drusen: The deposits associated with aging and age-related macular degeneration. Exp. Eye Res. 2004, 78, 243–256. [Google Scholar] [CrossRef]
- Lynn, S.A.; Keeling, E.; Munday, R.; Gabha, G.; Griffiths, H.; Lotery, A.J.; Ratnayaka, J.A. The complexities underlying age-related macular degeneration: Could amyloid β play an important role? Neural Regen. Res. 2017, 12, 538–548. [Google Scholar]
- Rivera, A.; White, K.; Stohr, H.; Steiner, K.; Hemmrich, N.; Grimm, T.; Jurklies, B.; Lorenz, B.; Scholl, H.P.; Apfelstedt-Sylla, E.; et al. A comprehensive survey of sequence variation in the ABCA4 (ABCR) gene in Stargardt disease and age-related macular degeneration. Am. J. Hum. Genet. 2000, 67, 800–813. [Google Scholar] [CrossRef] [Green Version]
- Wangtiraumnuay, N.; Capasso, J.; Tsukikawa, M.; Levin, A.; Biswas-Fiss, E. Novel ABCA4 mutation leads to loss of a conserved C-terminal motif: Implications for predicting pathogenicity based on genetic testing. Eur. J. Ophthalmol. 2018, 28, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.J.; Biswas, S.B.; Biswas-Fiss, E.E. Functional significance of the conserved C-terminal VFVNFA motif in the retina-specific ABC transporter, ABCA4, and its role in inherited visual disease. Biochem. Biophys. Res. Commun. 2019, 519, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.H.; Suh, S.; Foik, A.T.; Leinonen, H.; Newby, G.A.; Gao, X.D.; Banskota, S.; Hoang, T.; Du, S.W.; Dong, Z.; et al. In vivo base editing rescues cone photoreceptors in a mouse model of early-onset inherited retinal degeneration. Nat. Commun. 2022, 13, 1830. [Google Scholar] [CrossRef] [PubMed]
- Weber, B.H.; Lin, B.; White, K.; Kohler, K.; Soboleva, G.; Herterich, S.; Seeliger, M.W.; Jaissle, G.B.; Grimm, C.; Reme, C.; et al. A mouse model for Sorsby fundus dystrophy. Investig. Ophthalmol. Vis. Sci. 2002, 43, 2732–2740. [Google Scholar]
- Walton, R.T.; Christie, K.A.; Whittaker, M.N.; Kleinstiver, B.P. Unconstrained genome targeting with near-PAMless engineered CRISPR-Cas9 variants. Science 2020, 368, 290–296. [Google Scholar] [CrossRef]
- Vazquez-Dominguez, I.; Garanto, A.; Collin, R.W.J. Molecular Therapies for Inherited Retinal Diseases-Current Standing, Opportunities and Challenges. Genes 2019, 10, 654. [Google Scholar] [CrossRef] [Green Version]
- Trapani, I.; Colella, P.; Sommella, A.; Iodice, C.; Cesi, G.; de Simone, S.; Marrocco, E.; Rossi, S.; Giunti, M.; Palfi, A.; et al. Effective delivery of large genes to the retina by dual AAV vectors. EMBO Mol. Med. 2014, 6, 194–211. [Google Scholar] [CrossRef]
- Janssen, A.; Hoellenriegel, J.; Fogarasi, M.; Schrewe, H.; Seeliger, M.; Tamm, E.; Ohlmann, A.; May, C.A.; Weber, B.H.F.; Stöhr, H. Abnormal vessel formation in the choroid of mice lacking tissue inhibitor of metalloprotease-3. Investig. Ophthalmol. Vis. Sci. 2008, 49, 2812–2822. [Google Scholar] [CrossRef]
- Leco, K.J.; Waterhouse, P.; Sanchez, O.H.; Gowing, K.L.; Poole, A.R.; Wakeham, A.; Mak, T.W.; Khokha, R. Spontaneous air space enlargement in the lungs of mice lacking tissue inhibitor of metalloproteinases-3 (TIMP-3). J. Clin. Investig. 2001, 108, 817–829. [Google Scholar] [CrossRef]
- Kassiri, Z.; Oudit, G.Y.; Sanchez, O.; Dawood, F.; Mohammed, F.F.; Nuttall, R.K.; Etdwards, D.R.; Liu, P.P.; Balckx, P.H.; Khokha, R. Combination of tumor necrosis factor-α ablation and matrix metalloproteinase inhibition prevents heart failure after pressure overload in tissue inhibitor of metalloproteinase-3 knock-out mice. Circ. Res. 2005, 97, 380–390. [Google Scholar] [CrossRef] [Green Version]
- Mohammed, F.F.; Smookler, D.S.; Taylor, S.E.; Fingleton, B.; Kassiri, Z.; Sanchez, O.H.; English, J.L.; Matrisian, L.M.; Au, B.; Yeh, W.-C.; et al. Abnormal TNF activity in Timp3−/− mice leads to chronic hepatic inflammation and failure of liver regeneration. Nat. Genet. 2004, 36, 969–977. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Construct | Ideal Editing Window * | PAM Site * | Reference |
|---|---|---|---|
| SpCas9-ABE8e | 4′–8′ | 20′ NGG | [14] |
| SaCas9-ABE8e | 3′–14′ | 21′ NNGRRT | [14] |
| KKH-SaCas9-ABE8e | 3′–14′ | 21′ NNNRRT | [14] |
| CasMINI-ABE8e | 3′–4′ | Upstream of the target base: TTTR | [15] |
| NG-SpCas9-ABE | 4′–8′ | 20′NG | [16] |
| SpCas9-CBE | 4′–8′ | 20′ NGG | [17] |
| SaCas9-CBE | 2′–12′ | 21′ NNGRRT | [17] |
| SpCas9-GBE | 6′ | 20′ NGG | [18] |
| Option | Base Editor | Strand | Codon Change Patient > Edited | Amino Acid Change | SIFT | PolyPhen-2 Score | Mutation Taster Score |
|---|---|---|---|---|---|---|---|
| 1 | GBE | Reverse | TGC > TCC | p.Ser204Ser | Wild-type amino acid | Wild-type amino acid | Wild-type amino acid |
| 2 | GBE | Forward | TGC > TGG | p.Ser204Trp | Affects protein function, score = 0.01. Median sequence conservation: 3.37. | Possibly damaging with a score of 0.721 (sensitivity: 0.86; specificity: 0.92) | Polymorphism (Model: without_aae, prob: 0.999999181161545). Protein features (might be) affected—splice site changes. |
| 3 | ABE | Reverse | TGC > CGC | p.Ser204Arg | Tolerated, score = 0.06. Median sequence conservation: 3.37. | Benign, score = 0.023. Sensitivity: 0.95; specificity: 0.81. | Polymorphism (Model: without aae, prob: 0.999999621231165). |
| 4 | CBE | Forward | TGC > TGT | p.Ser204Cys | Identical to patient mutation | Identical to patient mutation | Identical to patient mutation |
| 5 | CBE | Reverse | TGC > TAC | p.Ser204Tyr | Affects protein function, score = 0.02. Median sequence conservation: 3.37. | Benign, score = 0.183. Sensitivity: 0.92; specificity: 0.87. | Polymorphism (Model: without_aae, prob: 0.999999618732328). Protein features (might be) affected—splice site changes |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elsayed, M.E.A.A.; Kaukonen, M.; Kiraly, P.; Kapetanovic, J.C.; MacLaren, R.E. Potential CRISPR Base Editing Therapeutic Options in a Sorsby Fundus Dystrophy Patient. Genes 2022, 13, 2103. https://doi.org/10.3390/genes13112103
Elsayed MEAA, Kaukonen M, Kiraly P, Kapetanovic JC, MacLaren RE. Potential CRISPR Base Editing Therapeutic Options in a Sorsby Fundus Dystrophy Patient. Genes. 2022; 13(11):2103. https://doi.org/10.3390/genes13112103
Chicago/Turabian StyleElsayed, Maram E. A. Abdalla, Maria Kaukonen, Peter Kiraly, Jasmina Cehajic Kapetanovic, and Robert E. MacLaren. 2022. "Potential CRISPR Base Editing Therapeutic Options in a Sorsby Fundus Dystrophy Patient" Genes 13, no. 11: 2103. https://doi.org/10.3390/genes13112103
APA StyleElsayed, M. E. A. A., Kaukonen, M., Kiraly, P., Kapetanovic, J. C., & MacLaren, R. E. (2022). Potential CRISPR Base Editing Therapeutic Options in a Sorsby Fundus Dystrophy Patient. Genes, 13(11), 2103. https://doi.org/10.3390/genes13112103