
Oculo-Cutaneous Albinism Type 4 (OCA4): Phenotype-Genotype Correlation

 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Methods
3. Results
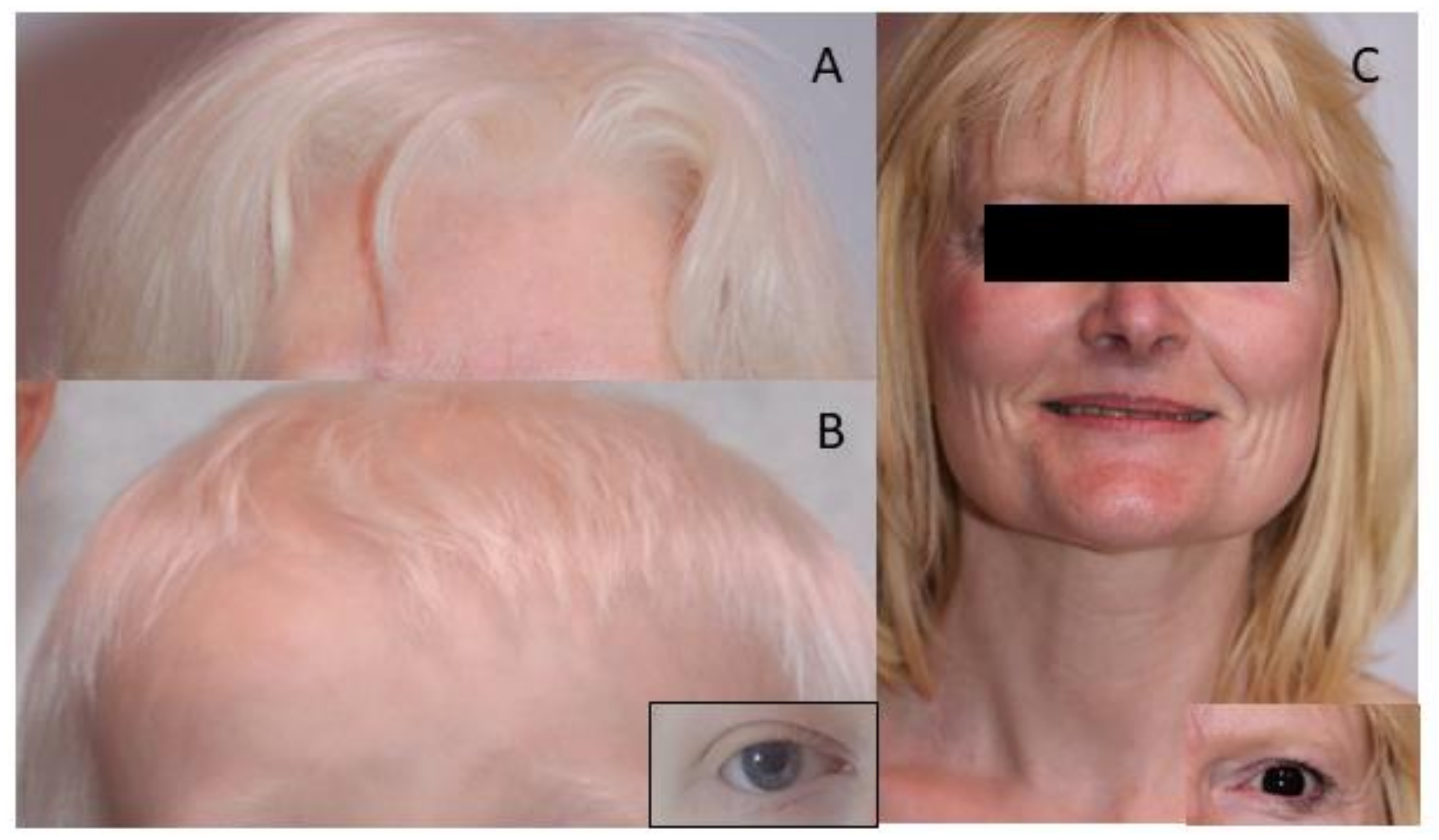
3.1. Dermatological Phenotype
3.2. Ocular Phenotype
3.3. Molecular Findings
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| OCA | Oculo-Cutaneous Albinism |
References
- Arveiler, B.; Lasseaux, E.; Morice-Picard, F. Clinical and genetic aspects of albinism. Presse Med. 2017, 46, 648–654. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Artero, E.; Morice-Picard, F.; Bremond-Gignac, D.; Drumare-Bouvet, I.; Duncombe-Poulet, C.; Leclerc-Mercier, S.; Dufresne, H.; Kaplan, J.; Jouanne, B.; Arveiler, B.; et al. Management of albinism: French guidelines for diagnosis and care. J. Eur. Acad. Dermatol. Venereol. 2021, 35, 1449–1459. [Google Scholar] [CrossRef] [PubMed]
- Al Abdi, L.; Alshammari, M.; Helaby, R.; Khan, A.O.; Alkuraya, F.S. PMEL is mutated in oculocutaneous albinism. Hum. Genet. 2022, 1–6. [Google Scholar]
- Grønskov, K.; Ek, J.; Brondum-Nielsen, K. Oculocutaneous albinism. Orphanet J. Rare Dis. 2007, 2, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marti, A.; Lasseaux, E.; Ezzedine, K.; Léauté-Labrèze, C.; Boralevi, F.; Paya, C.; Coste, V.; Deroissart, V.; Arveiler, B.; Taieb, A.; et al. Lessons of a day hospital: Comprehensive assessment of patients with albinism in a European setting. Pigment Cell Melanoma Res. 2018, 31, 318–329. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chi, W.; Tao, L.; Wang, G.; Deepak, R.N.V.K.; Sheng, L.; Chen, T.; Feng, Y.; Cao, X.; Cheng, L.; et al. Ablation of Proton/Glucose Exporter SLC45A2 Enhances Melanosomal Glycolysis to Inhibit Melanin Biosynthesis and Promote Melanoma Metastasis. J. Investig. Dermatol. 2022, 142, 2744–2755.e9. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, K.; Suzuki, T.; Shimizu, H.; Ishii, N.; Umezawa, Y.; Tada, J.; Kikuchi, N.; Takata, M.; Takamori, K.; Kishibe, M.; et al. Oculocutaneous albinism type 4 is one of the most common types of albinism in Japan. Am. J. Hum. Genet. 2004, 74, 466–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rundshagen, U.; Zühlke, C.; Opitz, S.; Schwinger, E.; Käsmann-Kellner, B. Mutations in the MATP gene in five German patients affected by oculocutaneous albinism type 4. Hum. Mutat. 2004, 23, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Lasseaux, E.; Plaisant, C.; Michaud, V.; Pennamen, P.; Trimouille, A.; Gaston, L.; Monfermé, S.; Lacombe, D.; Rooryck, C.; Morice-Picard, F.; et al. Molecular characterization of a series of 990 index patients with albinism. Pigment Cell Melanoma Res. 2018, 31, 466–474. [Google Scholar] [CrossRef] [PubMed]
- Rooryck, C.; Morice-Picard, F.; Elçioglu, N.H.; Lacombe, D.; Taieb, A.; Arveiler, B. Molecular diagnosis of oculocutaneous albinism: New mutations in the OCA1–4 genes and practical aspects. Pigment Cell Melanoma Res. 2018, 21, 583–587. [Google Scholar]
- Zhong, Z.; Gu, L.; Zheng, X.; Ma, N.; Wu, Z.; Duan, J.; Zhang, J.; Chen, J. Comprehensive analysis of spectral distribution of a large cohort of Chinese patients with non-syndromic oculocutaneous albinism facilitates genetic diagnosis. Pigment Cell Melanoma Res. 2019, 32, 672–686. [Google Scholar] [CrossRef] [PubMed]
- Shahzad, M.; University of Washington Center for Mendelian Genomics (UW CMG) Consortium; Yousaf, S.; Waryah, Y.M.; Gul, H.; Kausar, T.; Tariq, N.; Mahmood, U.; Ali, M.; Khan, M.A.; et al. Molecular outcomes, clinical consequences, and genetic diagnosis of Oculocutaneous Albinism in Pakistani population. Sci. Rep. 2017, 7, 44185. [Google Scholar] [CrossRef] [PubMed]
- Kruijt, C.C.; Schalij-Delfos, N.E.; de Wit, G.C.; Florijn, R.J.; van Genderen, M.M. Evident hypopigmentation without other ocular deficits in Dutch patients with oculocutaneous albinism type 4. Sci Rep. 2021, 11, 11572. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Patients | Gender, Age (Years) | Genetic Background | Consanguinity | Nevi | Eyes | Hair | Eyebrows Eyelashes | Nystagmus | Strabismus | VA | Refraction | ITI | MT | FHP | Variant 1 (SLC45A2 NM_016180.5) | Variant 2 (SLC45A2 NM_016180.5) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Group I | ||||||||||||||||
| P1 | M, 20 | Morocco | Yes | Present, amelanotic | Blue | White | White | Yes | Yes, esotropia | 1.6/10 RE; 2/10 LE | Hypermetropia astigmatism | NA | NA | Grade IV | NM_016180.5(SLC45A2):c.267_271del Chr5(GRCh37):g.33984422_33984426del p.(Ser90Glnfs*42) | NM_016180.5(SLC45A2):c.267_271del Chr5(GRCh37):g.33984422_33984426del p.(Ser90Glnfs*42) |
| P2 | F, 7 | Morocco | Yes | Present, pigmented | Blue | White blond | White | Yes | Yes, left exotropia | 1/20 RE; 1/20 LE | Hypermetropia Astigmatism | Grade IV | Grade II | Grade IV | NM_016180.5(SLC45A2):c.1028_1029del Chr5(GRCh37):g.33954469_33954470del p.(Gly343Alafs*10) | NM_016180.5(SLC45A2):c.1028_1029del Chr5(GRCh37):g.33954469_33954470del p.(Gly343Alafs*10) |
| P3 | M, 7 | Morocco | Yes | Present, pigmented | Blue | White blond | White | Yes | No | NA | Hypermetropia Astigmatism | Grade III | Grade II | Grade IV | NM_016180.5(SLC45A2):c.1028_1029del Chr5(GRCh37):g.33954469_33954470del p.(Gly343Alafs*10) | NM_016180.5(SLC45A2):c.1028_1029del Chr5(GRCh37):g.33954469_33954470del p.(Gly343Alafs*10) |
| P4 | F, 49 | France | No | Absent | Blue grey | White | White | Yes | No | 2/10 RE; 2/10 LE | NA | Grade IV | NA | Grade IV | NM_016180.5(SLC45A2):c.273del Chr5(GRCh37):g.33984417del p.(Ser92Alafs*21) | NM_016180.5(SLC45A2):c.1068C>G Chr5(GRCh37):g.33951747G>C p.(Asn356Lys) (N356K) |
| P5 | F, 63 | France | No | Present, amelanotic | Blue | White | White | Yes | No | 2/10 RE; 3/10 LE | NA | Grade III | NA | Grade III | NM_016180.5(SLC45A2):c.986del Chr5(GRCh37):g.33954512del p.(Thr329Lysfs*69) | NM_016180.5(SLC45A2):c.1036G>T Chr5(GRCh37):g.33951779C>A p.(Val346Leu) (V346L) |
| P6 | M, 18 | France | No | Present, pigmented | Blue | White | White | Yes | No | 1/10 RE; 1/10 LE | NA | Grade IV | NA | Grade IV | NM_016180.5(SLC45A2):c.986del Chr5(GRCh37):g.33954512del p.(Thr329Lysfs*69) | NM_016180.5(SLC45A2):c.1471G>A Chr5(GRCh37):g.33944875C>T p.(Gly491Arg) (G491R) |
| P7 | M, 9 | France | No | Absent | Blue | White | White | Yes | No | 1/10 RE; 1/10 LE | NA | Grade IV | NA | Grade IV | NM_016180.5(SLC45A2):c.986del Chr5(GRCh37):g.33954512del p.(Thr329Lysfs*69) | NM_016180.5(SLC45A2):c.1471G>A Chr5(GRCh37):g.33944875C>T p.(Gly491Arg) (G491R) |
| P8 | M, 7 | France | No | Absent | Blue grey | White | White | Yes | Yes, esotropia | 3/10 RE; 3/10 LE | NA | Grade III | NA | Grade IV | NM_016180.5(SLC45A2):c.986del Chr5(GRCh37):g.33954512del p.(Thr329Lysfs*69) | NM_016180.5(SLC45A2):c.1036G>T Chr5(GRCh37):g.33951779C>A p.(Val346Leu) (V346L) |
| P9 | M, 16 | France | No | Present, pigmented | Blue grey | White | White | Yes | No | 1/10 RE; 1/10 LE | NA | Grade IV | NA | Grade IV | NM_016180.5(SLC45A2):c.986del Chr5(GRCh37):g.33954512del p.(Thr329Lysfs*69) | NM_016180.5(SLC45A2):c.986del Chr5(GRCh37):g.33954512del p.(Thr329Lysfs*69) |
| P10 | F, 52 | France | No | Absent | Blue grey | White | White | Yes | Yes, esotropia | 1/10 RE; 1/10 LE | NA | Grade II | NA | Grade IV | NM_016180.5(SLC45A2):c.986del Chr5(GRCh37):g.33954512del p.(Thr329Lysfs*69) | NM_016180.5(SLC45A2):c.1166_1167del Chr5(GRCh37):g.33947470_33947471del p.(Lys389Serfs*55) |
| P11 | F, 50 | France | No | Present, amelanotic | Blue grey | White | White | Yes | No | 2/10 RE; 2/10 LE | NA | Grade IV | NA | Grade IV | NM_016180.5(SLC45A2):c.986del Chr5(GRCh37):g.33954512del p.(Thr329Lysfs*69) | NM_016180.5(SLC45A2):c.1166_1167del Chr5(GRCh37):g.33947470_33947471del p.(Lys389Serfs*55) |
| P12 | M, 65 | France | No | Present, amelanotic | Blue | White | White | Yes | Yes, esotropia | 2/10 RE; 2,5/10 LE | NA | Grade III | NA | Grade IV | NM_016180.5(SLC45A2):c.1273del Chr5(GRCh37):g.33947365del p.(Leu425Trpfs*9) | NM_016180.5(SLC45A2):c.1506del Chr5(GRCh37):g.33944842del p.(Thr503Profs*6) |
| P13 | F, 62 | France | No | Present, amelanotic | Blue | White | White | Yes | Yes, esotropia | 1.6/10 RE; 1.6/10 LE | NA | Grade III | NA | Grade IV | NM_016180.5(SLC45A2):c.1273del Chr5(GRCh37):g.33947365del p.(Leu425Trpfs*9) | NM_016180.5(SLC45A2):c.1506del Chr5(GRCh37):g.33944842del p.(Thr503Profs*6) |
| P14 | F, 38 | France | No | Present, amelanotic | Blue | White | White | Yes | Yes, esotropia | 1/10 RE; 1/10 LE | NA | Grade III | NA | Grade III | NM_016180.5(SLC45A2):c.147C>G Chr5(GRCh37):g.33984542G>C p.(Tyr49*) | NM_016180.5(SLC45A2):c.147C>G Chr5(GRCh37):g.33984542G>C p.(Tyr49*) |
| P15 | M, 42 | France | No | Present, amelanotic | Blue grey | White | White | Yes | Yes, esotropia | 2/10 RE; 2/10 LE | NA | Grade III | NA | Grade IV | NM_016180.5(SLC45A2):c.986del Chr5(GRCh37):g.33954512del p.(Thr329Lysfs*69) | NM_016180.5(SLC45A2):c.1466T>C Chr5(GRCh37):g.33944880A>G p.(Leu489Pro) (L489P) |
| P16 | F, 30 | France | Yes | Present, amelanotic | Blue | White | White | Yes | No | 1.6/10 RE; 2/10 LE | Hypermetropia Astigmatism | Grade IV | Grade II | Grade IV | NM_016180.5(SLC45A2):c.179T>G Chr5(GRCh37):g.33984510A>C p.(Leu60Arg) (L60R) | NM_016180.5(SLC45A2):c.179T>G Chr5(GRCh37):g.33984510A>C p.(Leu60Arg) (L60R) |
| P17 | F, 42 | France | No | Present, pigmented | Blue | White | White | Yes | Yes, esotropia | 1.6/10 RE; 2/10 LE | Hypermetropia Astigmatism | Grade IV | Grade II | Grade III | NM_016180.5(SLC45A2):c.986del Chr5(GRCh37):g.33954512del p.(Thr329Lysfs*69) | NM_016180.5(SLC45A2):c.1532C>A Chr5(GRCh37):g.33944814G>T p.(Ala511Glu) (A511E) |
| P18 | F, 66 | France | Yes | Present, amelanotic | Blue | White | White | Yes | Yes, exotropia | 1/10 RE; 1/10 LE | Hypermetropia Astigmatism | Grade III | Grade II | Grade III | NM_016180.5(SLC45A2):c.986del Chr5(GRCh37):g.33954512del p.(Thr329Lysfs*69) | NM_016180.5(SLC45A2):c.986del Chr5(GRCh37):g.33954512del p.(Thr329Lysfs*69) |
| P19 | M, 2 | France | No | Absent | Blue | White | White | Yes | No | NA | Grade III | Grade III | ND | NM_016180.5(SLC45A2):c.130G>A Chr5(GRCh37):g.33984559C>T p.(Gly44Arg) (G44R) | NM_016180.5(SLC45A2):c.986del Chr5(GRCh37):g.33954512del p.(Thr329Lysfs*69) | |
| P20 | F, 3 | France | No | Absent | Blue | White | White | Yes | No | NA | Grade IV | NA | Grade II | NM_016180.5(SLC45A2):c.1033-6_1033-3del Chr5(GRCh37):g.33951787_33951790del p.? | NM_016180.5(SLC45A2):c.986del Chr5(GRCh37):g.33954512del p.(Thr329Lysfs*69) | |
| Group 2 | ||||||||||||||||
| P21 | M, 11 | France | No | Present, pigmented | Blue | Blond | Blond | Yes | Yes, exotropia | 1.6/10 RE; 2/10 LE | Hypermetropia Astigmatism | NA | NA | NA | NM_016180.5(SLC45A2):c.533_534dup Chr5(GRCh37):g.33982369_33982370dup p.(Gly179Argfs*23) | NM_016180.5(SLC45A2):c.977T>A Chr5(GRCh37):g.33954521A>T p.(Ile326Asn) (I326N) |
| P22 | M, 7 | China | No | Present, pigmented | Brown | Dark blond | Brown | No | Yes, esotropia | 9/10 RE; 7/10 LE | Hypermetropia | No | Grade III | Grade I | NM_016180.5(SLC45A2):c.1045G>A Chr5(GRCh37):g.33951770C>T p.(Gly349Arg) (G349R) | NM_016180.5(SLC45A2):c.1255C>A Chr5(GRCh37):g.33947381G>T p.(Pro419Thr) (P419T) |
| P23 | M, 3 | France | No | Present, pigmented | Blue grey | Blond | Blond | Yes | No | 1/10 RE; 1/10 LE | Hypermetropia Astigmatism | Grade IV | Grade I | Grade IV | NM_016180.5(SLC45A2):c.1166_1167del Chr5(GRCh37):g.33947470_33947471del p.(Lys389Serfs*55) | NM_016180.5(SLC45A2):c.1471G>A Chr5(GRCh37):g.33944875C>T p.(Gly491Arg) (G491R) |
| P24 | F,10 | France | No | Absent | Blue | Blond | Blond | Yes | No | 2/10 RE; 2/10 LE | Hypermetropia | Grade II | NA | NA | Deletion exons 1-4 | NM_016180.5(SLC45A2):c.1518C>T Chr5(GRCh37):g.33944828G>A p.(Val506=) |
| P25 | F, 50 | France | No | Present, amelanotic | Blue grey | Red blond | Red blond | Yes | No | 3/10 RE; 3/10 LE | NA | Grade III | NA | Grade III | NM_016180.5(SLC45A2):c.606G>C Chr5(GRCh37):g.33964078C>G p.(Trp202Cys) (W202C) | NM_016180.5(SLC45A2):c.1532C>A Chr5(GRCh37):g.33944814G>T p.(Ala511Glu) (A511E) |
| P26 | F, 30 | France | No | Present, pigmented | Blue grey | Blond | White + Blond | Yes | No | 2/10 RE; 2/10 LE | Hypermetropia Astigmatism | No | Grade I | Grade IV | NM_016180.5(SLC45A2):c.258del Chr5(GRCh37):g.33984433del p.(Val87Trpfs*26) | NM_016180.5(SLC45A2):c.950A>G Chr5(GRCh37):g.33954548T>C p.(Tyr317Cys) (Y317C) |
| P27 | F, 27 | France | No | Present, pigmented | Blue | Blond | Blond | Yes | No | 2/10 RE; 2/10 LE | HypermetropiaAstigmatism | No | Grade I | Grade IV | NM_016180.5(SLC45A2):c.258del Chr5(GRCh37):g.33984433del p.(Val87Trpfs*26) | NM_016180.5(SLC45A2):c.950A>G Chr5(GRCh37):g.33954548T>C p.(Tyr317Cys) (Y317C) |
| P28 | F, 4 | France | No | Absent | Blue | Blond | Blond | No | No | NA | Hypermetropia Astigmatism | No | Grade II | Grade II | NM_016180.5(SLC45A2):c.953G>A Chr5(GRCh37):g.33954545C>T p.(Arg318His) (R318H) | NM_016180.5(SLC45A2):c.1518C>T Chr5(GRCh37):g.33944828G>A p.(Val506=) |
| P29 | F, 41 | Mauritania | No | Present, pigmented | Brown | Blond | Blond | Yes | Yes microexotropia | 1.2/10 RE; 1.4/10 LE | Myopia Astigmatism | Grade I | Grade III | Grade III | NM_016180.5(SLC45A2):c.1532C>T Chr5(GRCh37):g.33944814G>A p.(Ala511Val) (A511V) | NM_016180.5(SLC45A2):c.1532C>T Chr5(GRCh37):g.33944814G>A p.(Ala511Val) (A511V) |
| P30 | M, 30 | France | No | Present, pigmented | Blue | Blond | Blond | Yes | No | 5/10 RE; 5/10 LE | HypermetropiaAstigmatism | Grade I | NA | NA | NM_016180.5(SLC45A2):c.806G>A Chr5(GRCh37):g.33963878C>T p.(Gly269Asp) (G269D) | NM_016180.5(SLC45A2):c.1471G>A Chr5(GRCh37):g.33944875C>T p.(Gly491Arg) (G491R) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moreno-Artero, E.; Morice-Picard, F.; Lasseaux, E.; Robert, M.P.; Coste, V.; Michaud, V.; Leclerc-Mercier, S.; Bremond-Gignac, D.; Arveiler, B.; Hadj-Rabia, S. Oculo-Cutaneous Albinism Type 4 (OCA4): Phenotype-Genotype Correlation. Genes 2022, 13, 2198. https://doi.org/10.3390/genes13122198
Moreno-Artero E, Morice-Picard F, Lasseaux E, Robert MP, Coste V, Michaud V, Leclerc-Mercier S, Bremond-Gignac D, Arveiler B, Hadj-Rabia S. Oculo-Cutaneous Albinism Type 4 (OCA4): Phenotype-Genotype Correlation. Genes. 2022; 13(12):2198. https://doi.org/10.3390/genes13122198
Chicago/Turabian StyleMoreno-Artero, Ester, Fanny Morice-Picard, Eulalie Lasseaux, Matthieu P. Robert, Valentine Coste, Vincent Michaud, Stéphanie Leclerc-Mercier, Dominique Bremond-Gignac, Benoit Arveiler, and Smail Hadj-Rabia. 2022. "Oculo-Cutaneous Albinism Type 4 (OCA4): Phenotype-Genotype Correlation" Genes 13, no. 12: 2198. https://doi.org/10.3390/genes13122198
APA StyleMoreno-Artero, E., Morice-Picard, F., Lasseaux, E., Robert, M. P., Coste, V., Michaud, V., Leclerc-Mercier, S., Bremond-Gignac, D., Arveiler, B., & Hadj-Rabia, S. (2022). Oculo-Cutaneous Albinism Type 4 (OCA4): Phenotype-Genotype Correlation. Genes, 13(12), 2198. https://doi.org/10.3390/genes13122198