
Comparison of Metagenomics and Metatranscriptomics Tools: A Guide to Making the Right Choice

 ,
, 
Abstract
:1. Introduction
1.1. Microbiota: Human Health, Disease and Treatment
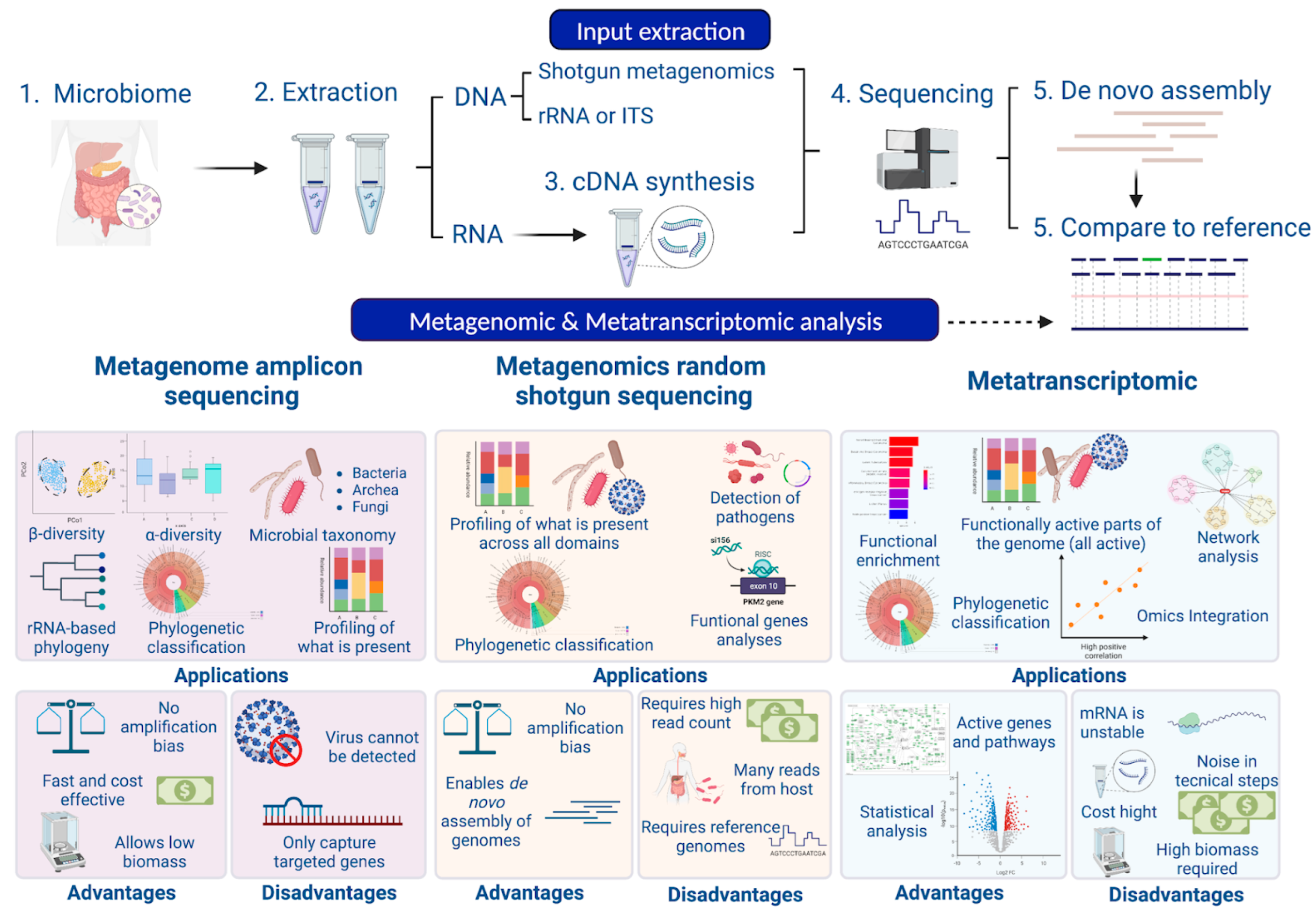
1.2. Techniques Based on Next-Generation Sequencing
1.3. Bioinformatic Tools
1.3.1. Pre-Processing
Trimming and Quality Filter
Host Removal
1.3.2. Taxonomic Identification
Assembly
Taxonomic Classification
Taxonomic Profiles Based on Marker Genes
Taxonomic Profiling Based on Whole Genomes and Transcriptomes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Preprocessing Tools | |||
|---|---|---|---|
| Step | Tools | Description | Ref |
| Quality report |
| Reads quality, seq length distribution and GC% | [86] |
| Summarise results | [87] | |
| Match a library with libraries expectation DB | [90] | |
| Trimming |
| Find and remove adapters, primers, poly-A tails and others | [88] |
| Trims and filters by k-mers and entropy and downsampling reads | [153] | |
| k-mer error trimming | [154] | |
| Downsampling reads | [155] | |
| Trimming tool for Illumina | [89] | |
| Host removal |
| Host sequences removal | [92] |
| Quality, trimming and host sequences removal | [93] | |
| Assembly for taxonomic classification | |||
| Assembly |
| Single k-mer Bruijn-graph-based assemblers | [100] |
| Single k-mer Bruijn-graph-based assemblers | [101] | |
| Multiple k-mers with preassembled at each interaction | [103] | |
| Multiple k-mers better assemblies with different abundances | [105] | |
| Iterative k-mer fast and co-assembly robust metagenomic tool | [107] | |
| Evaluate quality of assemblies and contamination | [152] | |
| Assess genome assembly and gene set completeness based on single-copy orthologs | [156] | |
| Taxonomic classification | |||
| Based on marker genes |
| Marker-gene-based taxonomic profiler | [157] |
| Taxonomic profiler based on a set of 40 prokaryotic marker genes | [120] | |
| Reads against unique subsequences at multiple taxonomic ranks | [121] | |
| MinHash-based taxonomic profiler enabling super-fast overlap estimations | [123] | |
| Dast searches with sequence bloom trees for taxonomic profiling | [124] | |
| Completely re-engineered microbiome platform based on QIIME | [69] | |
| Based on whole genomes and transcriptomes |
| Uses single-nucleotide polymorphism patterns | [140] |
| Uses the similarity in the genomic sequence and alignment tool | [141] | |
| Uses BLAST or DIAMOND to match sequences and assigns LCA of matches | [137] | |
| Genome Relative Abundance using Mixture Model theory | [138] | |
| Genome Relative Abundance in a non-negative LASSO approach | [139] | |
| k-mer search of reads against a DB built from multiple genomes | [142] | |
| k-mer search of reads against a DB built from completed genomes | [144] | |
| Pseudo-assembled using k-mers using DB of nonoverlapping | [158] | |
| Pseudo-assembled using k-mers using DB of nonoverlapping | [148] | |
| k-mer-based technique validated using the Smith–Waterman algorithm | [149] | |
| Spaced seeds with a reduced amino acid using protein homology search | [150] | |
| FM index using classifier against protein sequences with reduced amino acid | [151] | |
1.4. Pipelines for Metagenomics and Metatranscriptomics Analysis
2. Materials and Methods
2.1. Description of Three Datasets Used
2.1.1. 10/100/400 Species Next Generation Sequencing Datasets
2.1.2. Gut Microbiota Test Datasets
2.1.3. Description of Experimental Samples
2.2. Software and Databases Used in the Analysis
2.2.1. Kraken/Bracken/Krona
2.2.2. QIIME2
2.2.3. MetagenomeSeq
2.2.4. Nextflow
2.3. Dataset Processing
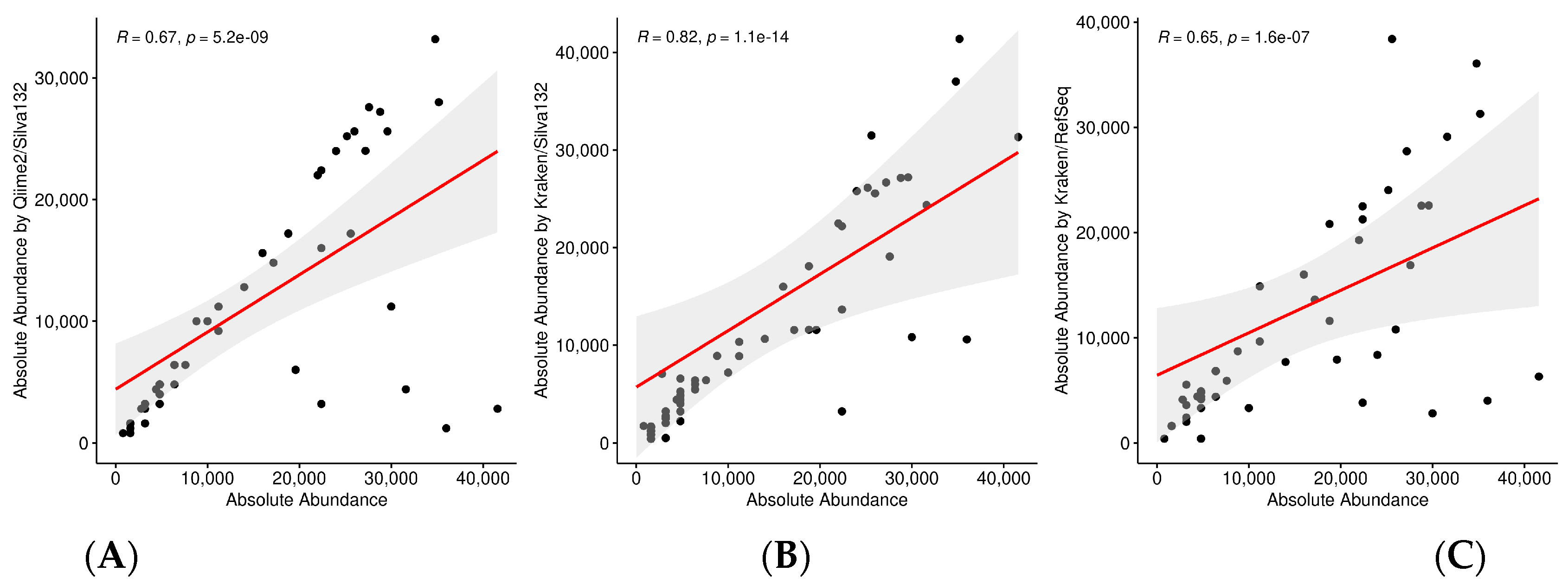
2.4. Correlation Analysis
3. Results
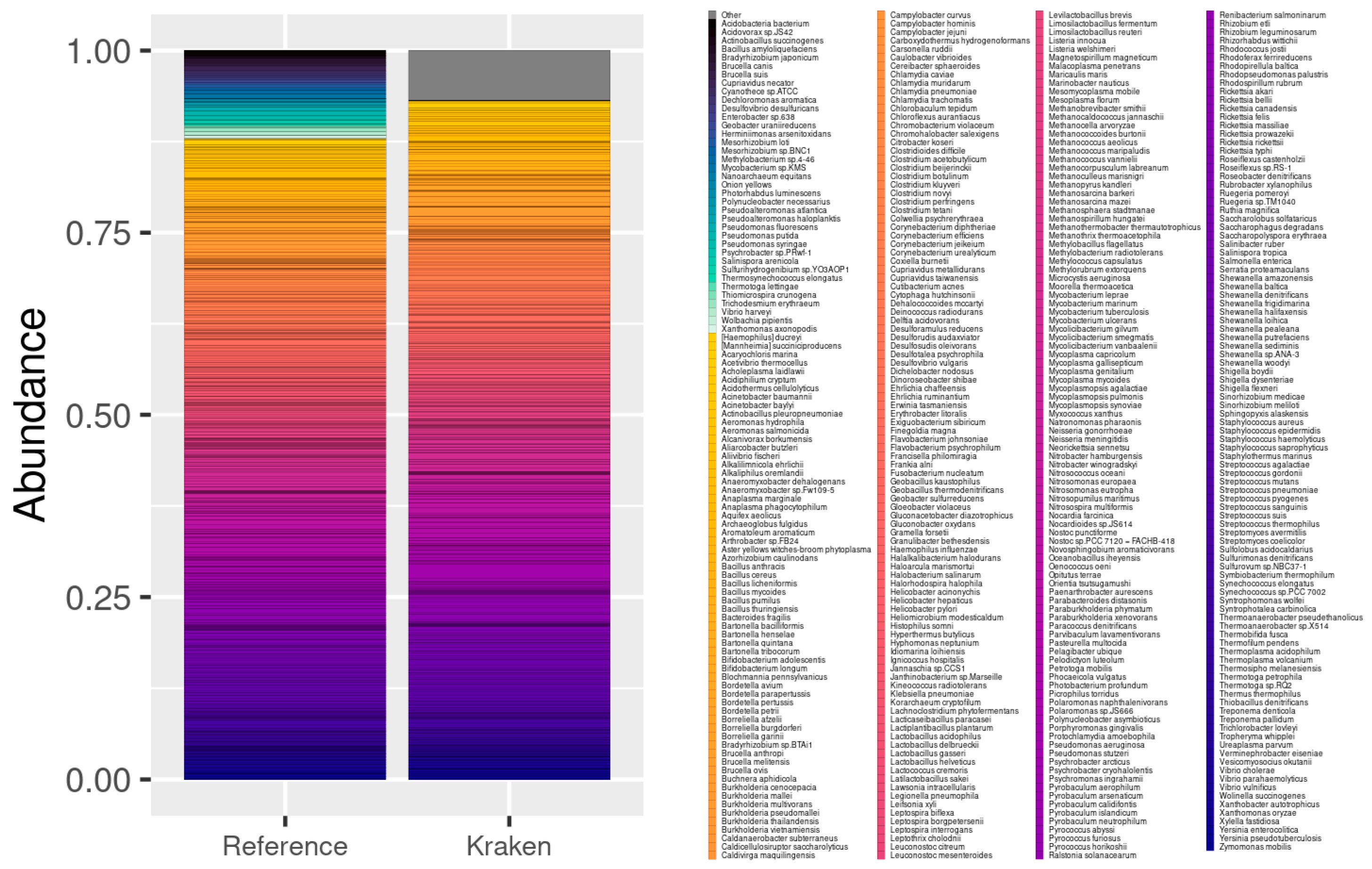
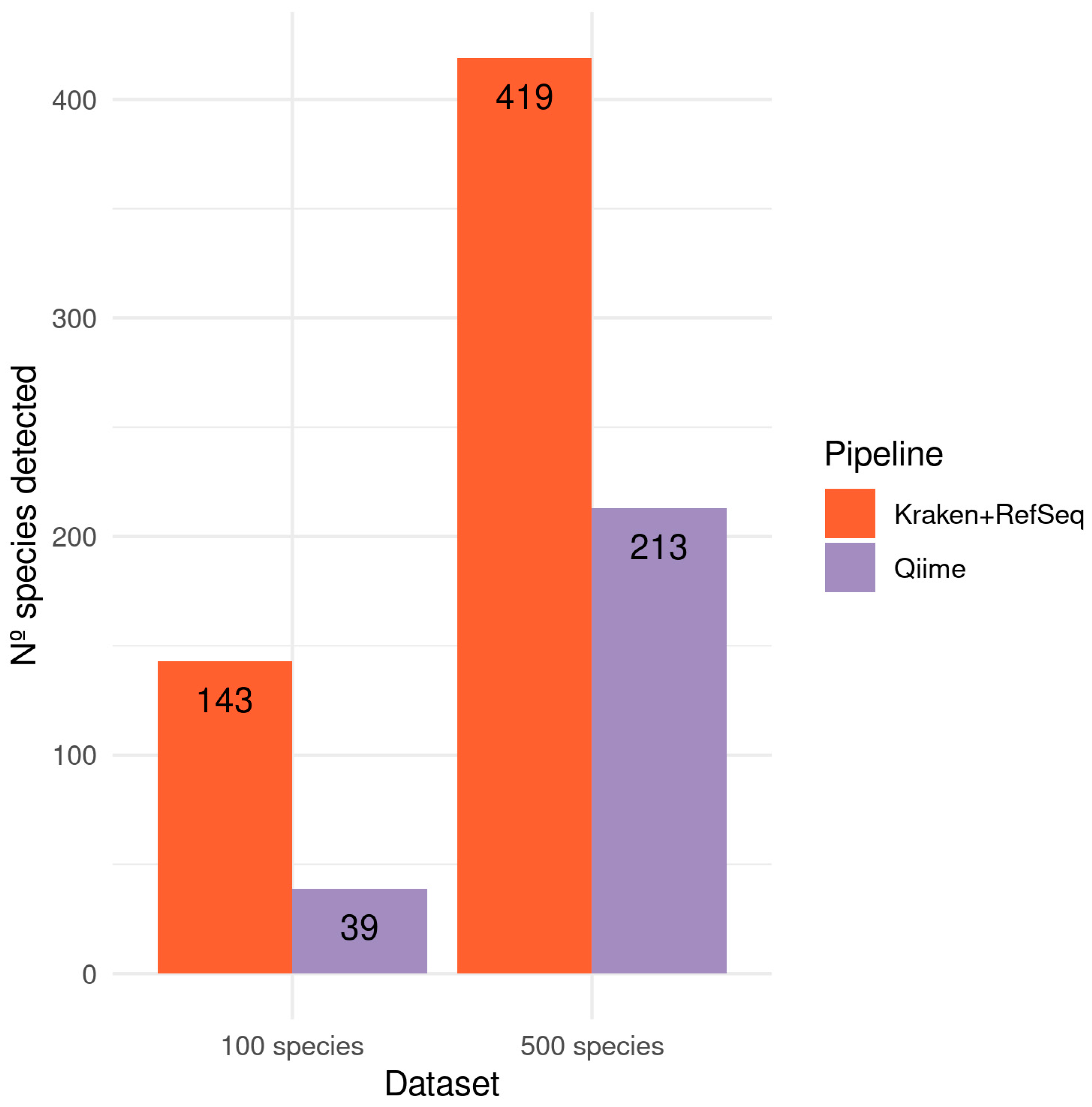
3.1. Simulated Samples
3.1.1. 10/100/400 Species Next Generation Sequencing Datasets
3.1.2. Gut Microbiota Test Datasets
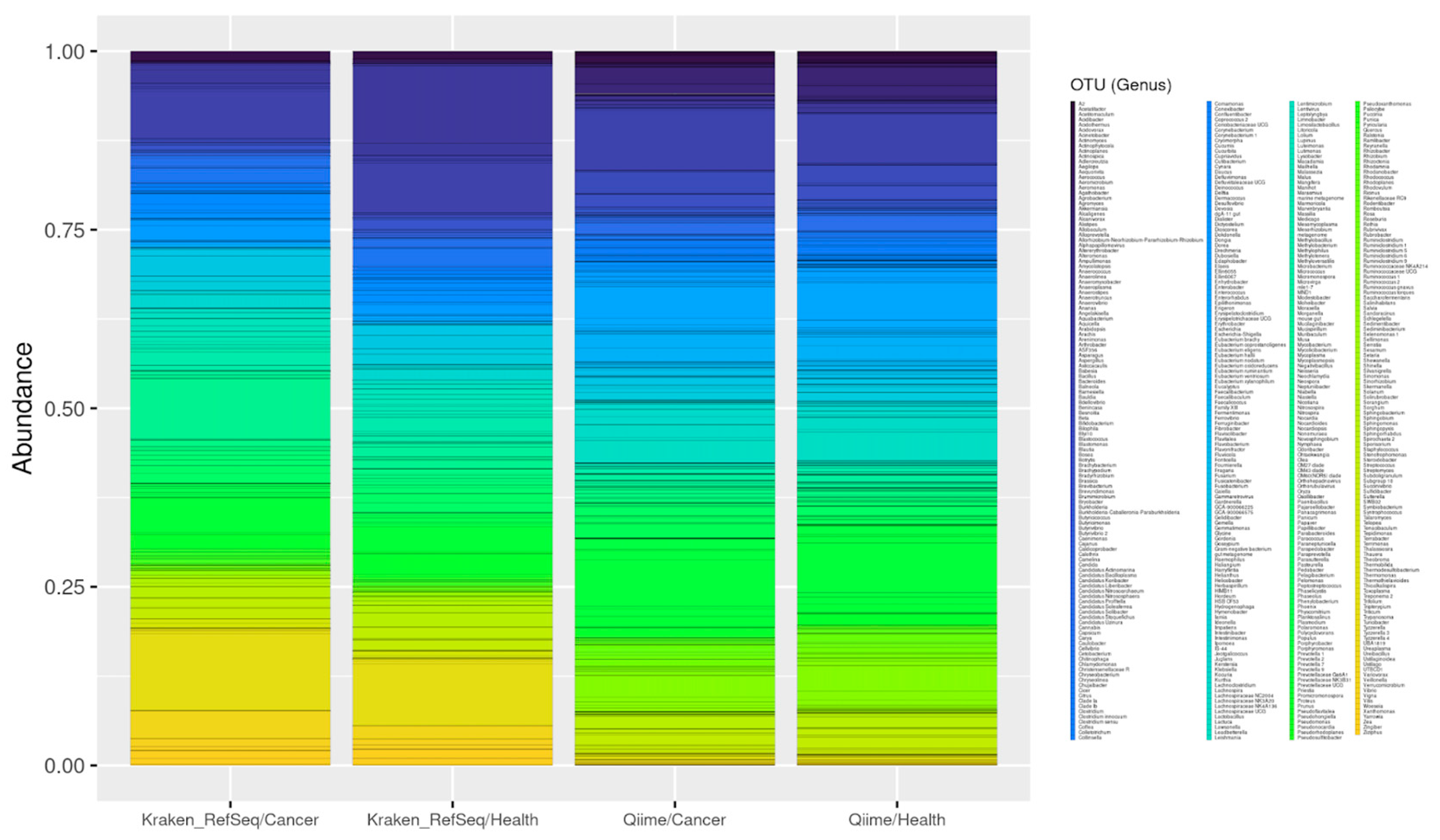
3.2. Case Study—Microorganisms Detected in Endometrial Cancer Samples
3.2.1. 16S Study
3.2.2. RNASeq Samples
3.3. Computational Times
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bianconi, E.; Piovesan, A.; Facchin, F.; Beraudi, A.; Casadei, R.; Frabetti, F.; Vitale, L.; Pelleri, M.C.; Tassani, S.; Piva, F.; et al. An estimation of the number of cells in the human body. Ann. Hum. Biol. 2013, 40, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Salzberg, S.L. Between a chicken and a grape: Estimating the number of human genes. Genome Biol. 2010, 11, 206. [Google Scholar] [CrossRef] [PubMed]
- Savage, D.C. Microbial Ecology of the Gastrointestinal Tract. Annu. Rev. Microbiol. 1977, 31, 107–133. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Ley, R.E.; Hamady, M.; Fraser-Liggett, C.M.; Knight, R.; Gordon, J.I. The Human Microbiome Project: Exploring the Microbial Part of Ourselves in a Changing World. Nature 2007, 449, 804–810. [Google Scholar] [CrossRef] [Green Version]
- Ravel, J.; Blaser, M.J.; Braun, J.; Brown, E.; Bushman, F.D.; Chang, E.B.; Davies, J.; Dewey, K.G.; Dinan, T.; Dominguez-Bello, M.; et al. Human Microbiome Science: Vision for the Future, Bethesda, MD, July 24 to 26, 2013. Microbiome 2014, 2, 16. [Google Scholar] [CrossRef] [Green Version]
- Clemente, J.C.; Manasson, J.; Scher, J.U. The role of the gut microbiome in systemic inflammatory disease. BMJ 2018, 360, j5145. [Google Scholar] [CrossRef]
- Sender, R.; Fuchs, S.; Milo, R. Are We Really Vastly Outnumbered? Revisiting the Ratio of Bacterial to Host Cells in Humans. Cell 2016, 164, 337–340. [Google Scholar] [CrossRef] [Green Version]
- Cani, P.D. Human gut microbiome: Hopes, threats and promises. Gut 2018, 67, 1716–1725. [Google Scholar] [CrossRef] [Green Version]
- Isaac, S.; Scher, J.U.; Djukovic, A.; Jiménez, N.; Littman, D.R.; Abramson, S.B.; Pamer, E.G.; Ubeda, C. Short- and long-term effects of oral vancomycin on the human intestinal microbiota. J. Antimicrob. Chemother. 2017, 72, 128–136. [Google Scholar] [CrossRef] [Green Version]
- Abdollahi-Roodsaz, S.; Abramson, S.B.; Scher, J.U. The metabolic role of the gut microbiota in health and rheumatic disease: Mechanisms and interventions. Nat. Rev. Rheumatol. 2016, 12, 446–455. [Google Scholar] [CrossRef]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef] [Green Version]
- Kernbauer, E.; Ding, Y.; Cadwell, K. An enteric virus can replace the beneficial function of commensal bacteria. Nature 2014, 516, 94–98. [Google Scholar] [CrossRef] [Green Version]
- Koren, O.; Goodrich, J.K.; Cullender, T.C.; Spor, A.; Laitinen, K.; Bäckhed, H.K.; Gonzalez, A.; Werner, J.J.; Angenent, L.T.; Knight, R.; et al. Host Remodeling of the Gut Microbiome and Metabolic Changes during Pregnancy. Cell 2012, 150, 470–480. [Google Scholar] [CrossRef] [Green Version]
- Pan, K.; Zhang, C.; Tian, J. The Effects of Different Modes of Delivery on the Structure and Predicted Function of Intestinal Microbiota in Neonates and Early Infants. Pol. J. Microbiol. 2021, 70, 45–55. [Google Scholar] [CrossRef]
- Boudar, Z.; Sehli, S.; El Janahi, S.; Al Idrissi, N.; Hamdi, S.; Dini, N.; Brim, H.; Amzazi, S.; Nejjari, C.; Lloyd-Puryear, M.; et al. Metagenomics Approaches to Investigate the Neonatal Gut Microbiome. Front. Pediatr. 2022, 10, 886627. [Google Scholar] [CrossRef]
- Shamriz, O.; Mizrahi, H.; Werbner, M.; Shoenfeld, Y.; Avni, O.; Koren, O. Microbiota at the crossroads of autoimmunity. Autoimmun. Rev. 2016, 15, 859–869. [Google Scholar] [CrossRef]
- Arrieta, M.-C.; Stiemsma, L.T.; Amenyogbe, N.; Brown, E.M.; Finlay, B. The Intestinal Microbiome in Early Life: Health and Disease. Front. Immunol. 2014, 5, 427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhuang, L.; Chen, H.; Zhang, S.; Zhuang, J.; Li, Q.; Feng, Z. Intestinal Microbiota in Early Life and Its Implications on Childhood Health. Genom. Proteom. Bioinform. 2019, 17, 13–25. [Google Scholar] [CrossRef]
- Adak, A.; Khan, M.R. An insight into gut microbiota and its functionalities. Cell. Mol. Life Sci. 2019, 76, 473–493. [Google Scholar] [CrossRef] [PubMed]
- Torres-Fuentes, C.; Schellekens, H.; Dinan, T.G.; Cryan, J.F. The microbiota–gut–brain axis in obesity. Lancet Gastroenterol. Hepatol. 2017, 2, 747–756. [Google Scholar] [CrossRef] [PubMed]
- Gomaa, E.Z. Human gut microbiota/microbiome in health and diseases: A review. Antonie Van Leeuwenhoek 2020, 113, 2019–2040. [Google Scholar] [CrossRef]
- Dalby, M.J.; Hall, L.J. Recent advances in understanding the neonatal microbiome. F1000Research 2020, 9, 422. [Google Scholar] [CrossRef] [PubMed]
- Tamburini, S.; Shen, N.; Wu, H.C.; Clemente, J.C. The Microbiome in Early Life: Implications for Health Outcomes. Nat. Med. 2016, 22, 713–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dominguez-Bello, M.G.; Godoy-Vitorino, F.; Knight, R.; Blaser, M.J. Role of the microbiome in human development. Gut 2019, 68, 1108–1114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oniszczuk, A.; Oniszczuk, T.; Gancarz, M.; Szymańska, J. Role of Gut Microbiota, Probiotics and Prebiotics in the Cardiovascular Diseases. Molecules 2021, 26, 1172. [Google Scholar] [CrossRef] [PubMed]
- Guan, Q. A Comprehensive Review and Update on the Pathogenesis of Inflammatory Bowel Disease. J. Immunol. Res. 2019, 2019, 7247238. [Google Scholar] [CrossRef] [Green Version]
- Shan, Y.; Lee, M.; Chang, E.B. The Gut Microbiome and Inflammatory Bowel Diseases. Annu. Rev. Med. 2022, 73, 455–468. [Google Scholar] [CrossRef]
- De Luca, F.; Shoenfeld, Y. The microbiome in autoimmune diseases. Clin. Exp. Immunol. 2019, 195, 74–85. [Google Scholar] [CrossRef] [Green Version]
- De Oliveira, G.L.V.; Leite, A.Z.; Higuchi, B.S.; Gonzaga, M.I.; Mariano, V.S. Intestinal dysbiosis and probiotic applications in autoimmune diseases. Immunology 2017, 152, 1–12. [Google Scholar] [CrossRef]
- Socała, K.; Doboszewska, U.; Szopa, A.; Serefko, A.; Włodarczyk, M.; Zielińska, A.; Poleszak, E.; Fichna, J.; Wlaź, P. The role of microbiota-gut-brain axis in neuropsychiatric and neurological disorders. Pharmacol. Res. 2021, 172, 105840. [Google Scholar] [CrossRef]
- Cryan, J.F.; O’Riordan, K.J.; Cowan, C.S.M.; Sandhu, K.V.; Bastiaanssen, T.F.S.; Boehme, M.; Codagnone, M.G.; Cussotto, S.; Fulling, C.; Golubeva, A.V.; et al. The Microbiota-Gut-Brain Axis. Physiol. Rev. 2019, 99, 1877–2013. [Google Scholar] [CrossRef] [PubMed]
- Garrett, W.S. Cancer and the microbiota. Science 2015, 348, 80–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matson, V.; Chervin, C.S.; Gajewski, T.F. Cancer and the Microbiome—Influence of the Commensal Microbiota on Cancer, Immune Responses, and Immunotherapy. Gastroenterology 2021, 160, 600–613. [Google Scholar] [CrossRef] [PubMed]
- Kogut, M.H.; Lee, A.; Santin, E. Microbiome and pathogen interaction with the immune system. Poult. Sci. 2020, 99, 1906–1913. [Google Scholar] [CrossRef]
- Chen, H.; Li, H.; Liu, Z. Interplay of intestinal microbiota and mucosal immunity in inflammatory bowel disease: A relationship of frenemies. Ther. Adv. Gastroenterol. 2020, 13, 1756284820935188. [Google Scholar] [CrossRef]
- Chunxi, L.; HaiYue, L.; Yanxia, L.; Jianbing, P.; Jin, S. The Gut Microbiota and Respiratory Diseases: New Evidence. J. Immunol. Res. 2020, 2020, 2340670. [Google Scholar] [CrossRef]
- Chioma, O.S.; Hesse, L.E.; Chapman, A.; Drake, W.P. Role of the Microbiome in Interstitial Lung Diseases. Front. Med. 2021, 8, 595522. [Google Scholar] [CrossRef]
- Maeda, Y.; Kurakawa, T.; Umemoto, E.; Motooka, D.; Ito, Y.; Gotoh, K.; Hirota, K.; Matsushita, M.; Furuta, Y.; Narazaki, M.; et al. Dysbiosis Contributes to Arthritis Development via Activation of Autoreactive T Cells in the Intestine. Arthritis Rheumatol. 2016, 68, 2646–2661. [Google Scholar] [CrossRef]
- Scher, J.U.; Sczesnak, A.; Longman, R.S.; Segata, N.; Ubeda, C.; Bielski, C.; Rostron, T.; Cerundolo, V.; Pamer, E.G.; Abramson, S.B.; et al. Expansion of intestinal Prevotella copri correlates with enhanced susceptibility to arthritis. eLife 2013, 2, e01202. [Google Scholar] [CrossRef]
- Alipour, B.; Homayouni-Rad, A.; Vaghef-Mehrabany, E.; Sharif, S.K.; Vaghef-Mehrabany, L.; Asghari-Jafarabadi, M.; Nakhjavani, M.R.; Mohtadi-Nia, J. Effects of Lactobacillus casei supplementation on disease activity and inflammatory cytokines in rheumatoid arthritis patients: A randomized double-blind clinical trial. Int. J. Rheum. Dis. 2014, 17, 519–527. [Google Scholar] [CrossRef]
- Zamani, B.; Golkar, H.R.; Farshbaf, S.; Emadi-Baygi, M.; Tajabadi-Ebrahimi, M.; Jafari, P.; Akhavan, R.; Taghizadeh, M.; Memarzadeh, M.R.; Asemi, Z. Clinical and metabolic response to probiotic supplementation in patients with rheumatoid arthritis: A randomized, double-blind, placebo-controlled trial. Int. J. Rheum. Dis. 2016, 19, 869–879. [Google Scholar] [CrossRef] [PubMed]
- Hatakka, K.; Martio, J.; Korpela, M.; Herranen, M.; Poussa, T.; Laasanen, T.; Saxelin, M.; Vapaatalo, H.; Moilanen, E.; Korpela, R. Effects of probiotic therapy on the activity and activation of mild rheumatoid arthritis—A pilot study. Scand. J. Rheumatol. 2003, 32, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Agus, A.; Planchais, J.; Sokol, H. Gut Microbiota Regulation of Tryptophan Metabolism in Health and Disease. Cell Host Microbe 2018, 23, 716–724. [Google Scholar] [CrossRef] [Green Version]
- Kang, D.-W.; Adams, J.B.; Coleman, D.M.; Pollard, E.L.; Maldonado, J.; McDonough-Means, S.; Caporaso, J.G.; Krajmalnik-Brown, R. Long-term benefit of Microbiota Transfer Therapy on autism symptoms and gut microbiota. Sci. Rep. 2019, 9, 5821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, N.; Chen, H.; Cheng, Y.; Xu, F.; Ruan, G.; Ying, S.; Tang, W.; Chen, L.; Chen, M.; Lv, L.; et al. Fecal Microbiota Transplantation Relieves Gastrointestinal and Autism Symptoms by Improving the Gut Microbiota in an Open-Label Study. Front. Cell. Infect. Microbiol. 2021, 11, 759435. [Google Scholar] [CrossRef] [PubMed]
- Dohlman, A.B.; Klug, J.; Mesko, M.; Gao, I.H.; Lipkin, S.M.; Shen, X.; Iliev, I.D. A pan-cancer mycobiome analysis reveals fungal involvement in gastrointestinal and lung tumors. Cell 2022, 185, 3807.e12–3822.e12. [Google Scholar] [CrossRef]
- Narunsky-Haziza, L.; Sepich-Poore, G.D.; Livyatan, I.; Asraf, O.; Martino, C.; Nejman, D.; Gavert, N.; Stajich, J.E.; Amit, G.; González, A.; et al. Pan-cancer analyses reveal cancer-type-specific fungal ecologies and bacteriome interactions. Cell 2022, 185, 3789.e17–3806.e17. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Ren, Z.; Gao, X.; Hu, X.; Zhou, Y.; Jiang, J.; Lu, H.; Yin, S.; Ji, J.; Zhou, L.; et al. Integrated analysis of microbiome and host transcriptome reveals correlations between gut microbiota and clinical outcomes in HBV-related hepatocellular carcinoma. Genome Med. 2020, 12, 102. [Google Scholar] [CrossRef] [PubMed]
- Sobhani, I.; Bergsten, E.; Couffin, S.; Amiot, A.; Nebbad, B.; Barau, C.; De’Angelis, N.; Rabot, S.; Canoui-Poitrine, F.; Mestivier, D.; et al. Colorectal cancer-associated microbiota contributes to oncogenic epigenetic signatures. Proc. Natl. Acad. Sci. USA 2019, 116, 24285–24295. [Google Scholar] [CrossRef] [PubMed]
- Witkowski, M.; Weeks, T.L.; Hazen, S.L. Gut Microbiota and Cardiovascular Disease. Circ. Res. 2020, 127, 553–570. [Google Scholar] [CrossRef]
- Nemet, I.; Saha, P.P.; Gupta, N.; Zhu, W.; Romano, K.A.; Skye, S.M.; Cajka, T.; Mohan, M.L.; Li, L.; Wu, Y.; et al. A Cardiovascular Disease-Linked Gut Microbial Metabolite Acts via Adrenergic Receptors. Cell 2020, 180, 862.e22–877.e22. [Google Scholar] [CrossRef]
- Yuan, J.; Chen, C.; Cui, J.; Lu, J.; Yan, C.; Wei, X.; Zhao, X.; Li, N.; Li, S.; Xue, G.; et al. Fatty Liver Disease Caused by High-Alcohol-Producing Klebsiella pneumoniae. Cell Metab. 2019, 30, 675.e7–688.e7. [Google Scholar] [CrossRef]
- NIH HMP Working Group; Peterson, J.; Garges, S.; Giovanni, M.; McInnes, P.; Wang, L.; Schloss, J.A.; Bonazzi, V.; McEwen, J.E.; Wetterstrand, K.A.; et al. The NIH Human Microbiome Project. Genome Res. 2009, 19, 2317–2323. [Google Scholar] [CrossRef] [Green Version]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marrazzo, J.M.; Martin, D.H.; Watts, D.H.; Schulte, J.; Sobel, J.D.; Hillier, S.L.; Deal, C.; Fredricks, D.N. Bacterial Vaginosis: Identifying Research Gaps Proceedings of a Workshop Sponsored by DHHS/NIH/NIAID. Sex. Transm. Dis. 2010, 37, 732–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turnbaugh, P.; Gordon, J.I. The core gut microbiome, energy balance and obesity. J. Physiol. 2009, 587, 4153–4158. [Google Scholar] [CrossRef] [PubMed]
- The Human Microbiome Project Consortium. A Framework for Human Microbiome Research. Nature 2012, 486, 215–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huttenhower, D.; Gevers, D.; Knight, R.; Abubucker, S.; White, O. The Human Microbiome Project Consortium Structure, Function and Diversity of the Healthy Human Microbiome. Nature 2012, 486, 207–214. [Google Scholar] [CrossRef] [Green Version]
- Aguiar-Pulido, V.; Huang, W.; Suarez-Ulloa, V.; Cickovski, T.; Mathee, K.; Narasimhan, G. Metagenomics, Metatranscriptomics, and Metabolomics Approaches for Microbiome Analysis. Evol. Bioinform. 2016, 12, 5–16. [Google Scholar] [CrossRef] [Green Version]
- The Integrative HMP (iHMP) Research Network Consortium. The Integrative Human Microbiome Project: Dynamic Analysis of Microbiome-Host Omics Profiles during Periods of Human Health and Disease. Cell Host Microbe 2014, 16, 276–289. [Google Scholar] [CrossRef]
- Welcome to The IHMP Data Portal|HMP. Available online: https://portal.hmpdacc.org/ (accessed on 17 October 2022).
- Dusko Ehrlich, S. MetaHIT consortium [Metagenomics of the intestinal microbiota: Potential applications]. Gastroenterol. Clin. Biol. 2010, 34, S23–S28. [Google Scholar] [CrossRef] [PubMed]
- Goedert, J.J.; Hua, X.; Yu, G.; Shi, J. Diversity and Composition of the Adult Fecal Microbiome Associated with History of Cesarean Birth or Appendectomy: Analysis of the American Gut Project. eBioMedicine 2014, 1, 167–172. [Google Scholar] [CrossRef] [Green Version]
- Sola-Leyva, A.; Andrés-León, E.; Molina, N.M.; Terron-Camero, L.C.; Plaza-Díaz, J.; Sáez-Lara, M.J.; Gonzalvo, M.C.; Sánchez, R.; Ruíz, S.; Martínez, L.; et al. Mapping the entire functionally active endometrial microbiota. Hum. Reprod. 2021, 36, 1021–1031. [Google Scholar] [CrossRef]
- Breitwieser, F.P.; Lu, J.; Salzberg, S.L. A review of methods and databases for metagenomic classification and assembly. Brief. Bioinform. 2019, 20, 1125–1136. [Google Scholar] [CrossRef] [PubMed]
- Abellan-Schneyder, I.; Matchado, M.S.; Reitmeier, S.; Sommer, A.; Sewald, Z.; Baumbach, J.; List, M.; Neuhaus, K. Primer, Pipelines, Parameters: Issues in 16S rRNA Gene Sequencing. Msphere 2021, 6, e01202-20. [Google Scholar] [CrossRef] [PubMed]
- Karst, S.M.; Dueholm, M.S.; McIlroy, S.J.; Kirkegaard, R.H.; Nielsen, P.H.; Albertsen, M. Retrieval of a million high-quality, full-length microbial 16S and 18S rRNA gene sequences without primer bias. Nat. Biotechnol. 2018, 36, 190–195. [Google Scholar] [CrossRef]
- Janda, J.M.; Abbott, S.L. 16S rRNA Gene Sequencing for Bacterial Identification in the Diagnostic Laboratory: Pluses, Perils, and Pitfalls. J. Clin. Microbiol. 2007, 45, 2761–2764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, Interactive, Scalable and Extensible Microbiome Data Science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Peters, D.L.; Wang, W.; Zhang, X.; Ning, Z.; Mayne, J.; Figeys, D. Metaproteomic and Metabolomic Approaches for Characterizing the Gut Microbiome. PROTEOMICS 2019, 19, e1800363. [Google Scholar] [CrossRef] [PubMed]
- Eloe-Fadrosh, E.A.; Paez-Espino, D.; Jarett, J.; Dunfield, P.; Hedlund, B.P.; Dekas, A.E.; Grasby, S.E.; Brady, A.L.; Dong, H.; Briggs, B.R.; et al. Global metagenomic survey reveals a new bacterial candidate phylum in geothermal springs. Nat. Commun. 2016, 7, 10476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, C.T.; Hug, L.A.; Thomas, B.C.; Sharon, I.; Castelle, C.J.; Singh, A.; Wilkins, M.J.; Wrighton, K.C.; Williams, K.H.; Banfield, J.F. Unusual biology across a group comprising more than 15% of domain Bacteria. Nature 2015, 523, 208–211. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Wang, K.; Wu, W.; Giannoulatou, E.; Ho, J.W.K.; Li, L. Host and microbiome multi-omics integration: Applications and methodologies. Biophys. Rev. 2019, 11, 55–65. [Google Scholar] [CrossRef]
- Shin, J.; Lee, S.; Go, M.-J.; Lee, S.Y.; Kim, S.C.; Lee, C.-H.; Cho, B.-K. Analysis of the mouse gut microbiome using full-length 16S rRNA amplicon sequencing. Sci. Rep. 2016, 6, 29681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Didelot, X.; Walker, A.S.; Peto, T.E.; Crook, D.W.; Wilson, D.J. Within-host evolution of bacterial pathogens. Nat. Rev. Genet. 2016, 14, 150–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharpton, T.J. An introduction to the analysis of shotgun metagenomic data. Front. Plant Sci. 2014, 5, 209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.-X.; Qin, Y.; Chen, T.; Lu, M.; Qian, X.; Guo, X.; Bai, Y. A practical guide to amplicon and metagenomic analysis of microbiome data. Protein Cell 2021, 12, 315–330. [Google Scholar] [CrossRef]
- Salazar, G.; Paoli, L.; Alberti, A.; Huerta-Cepas, J.; Ruscheweyh, H.-J.; Cuenca, M.; Field, C.M.; Coelho, L.P.; Cruaud, C.; Engelen, S.; et al. Gene Expression Changes and Community Turnover Differentially Shape the Global Ocean Metatranscriptome. Cell 2019, 179, 1068.e21–1083.e21. [Google Scholar] [CrossRef] [Green Version]
- Turner, T.R.; Ramakrishnan, K.; Walshaw, J.; Heavens, D.; Alston, M.; Swarbreck, D.; Osbourn, A.; Grant, A.; Poole, P.S. Comparative metatranscriptomics reveals kingdom level changes in the rhizosphere microbiome of plants. ISME J. 2013, 7, 2248–2258. [Google Scholar] [CrossRef] [Green Version]
- Sharpton, T.J. Role of the Gut Microbiome in Vertebrate Evolution. mSystems 2018, 3, e00174-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jovel, J.; Nimaga, A.; Jordan, T.; O’Keefe, S.; Patterson, J.; Thiesen, A.; Hotte, N.; Bording-Jorgensen, M.; Subedi, S.; Hamilton, J.; et al. Metagenomics Versus Metatranscriptomics of the Murine Gut Microbiome for Assessing Microbial Metabolism During Inflammation. Front. Microbiol. 2022, 13, 119. [Google Scholar] [CrossRef]
- Wilmes, P.; Heintz-Buschart, A.; Bond, P.L. A decade of metaproteomics: Where we stand and what the future holds. Proteomics 2015, 15, 3409–3417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldrian, P. Distribution of Extracellular Enzymes in Soils: Spatial Heterogeneity and Determining Factors at Various Scales. Soil Sci. Soc. Am. J. 2014, 78, 11–18. [Google Scholar] [CrossRef] [Green Version]
- Daliri, E.; Ofosu, F.; Chelliah, R.; Lee, B.; Oh, D.-H. Challenges and Perspective in Integrated Multi-Omics in Gut Microbiota Studies. Biomolecules 2021, 11, 300. [Google Scholar] [CrossRef] [PubMed]
- Sequencing and beyond: Integrating Molecular “Omics” for Microbial Community Profiling—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/25915636/ (accessed on 17 October 2022).
- Babraham Bioinformatics-FastQC a Quality Control Tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 17 October 2022).
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [Green Version]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Babraham Bioinformatics-FastQ Screen. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastq_screen/ (accessed on 17 October 2022).
- Bushnell, B.; Rood, J.; Singer, E. BBMerge—Accurate paired shotgun read merging via overlap. PLoS ONE 2017, 12, e0185056. [Google Scholar] [CrossRef] [PubMed]
- Mclver, L.J. BioBakery Workflow 2018. Available online: https://github.com/biobakery/biobakery_workflows (accessed on 28 November 2022).
- Andrés-León, E.; Núñez-Torres, R.; Rojas, A.M. miARma-Seq: A comprehensive tool for miRNA, mRNA and circRNA analysis. Sci. Rep. 2016, 6, 25749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrés-León, E.; Rojas, A.M. miARma-Seq, a comprehensive pipeline for the simultaneous study and integration of miRNA and mRNA expression data. Methods 2019, 152, 31–40. [Google Scholar] [CrossRef]
- Sangwan, N.; Xia, F.; Gilbert, J.A. Recovering complete and draft population genomes from metagenome datasets. Microbiome 2016, 4, 8. [Google Scholar] [CrossRef]
- Mande, S.S.; Mohammed, M.H.; Ghosh, T. Classification of metagenomic sequences: Methods and challenges. Brief. Bioinform. 2012, 13, 669–681. [Google Scholar] [CrossRef] [Green Version]
- Ghurye, J.S.; Cepeda-Espinoza, V.; Pop, M. Metagenomic Assembly: Overview, Challenges and Applications. Yale J. Biol. Med. 2016, 89, 353–362. [Google Scholar]
- Ayling, M.; Clark, M.D.; Leggett, R.M. New approaches for metagenome assembly with short reads. Brief. Bioinform. 2020, 21, 584–594. [Google Scholar] [CrossRef] [Green Version]
- Namiki, T.; Hachiya, T.; Tanaka, H.; Sakakibara, Y. MetaVelvet: An extension of Velvet assembler to de novo metagenome assembly from short sequence reads. Nucleic Acids Res. 2012, 40, e155. [Google Scholar] [CrossRef] [Green Version]
- Afiahayati, A.; Sato, K.; Sakakibara, Y. MetaVelvet-SL: An extension of the Velvet assembler to a de novo metagenomic assembler utilizing supervised learning. DNA Res. Int. J. Rapid Publ. Rep. Genes Genomes 2015, 22, 69–77. [Google Scholar] [CrossRef] [Green Version]
- Boisvert, S.; Raymond, F.; Godzaridis, é.; LaViolette, F.; Corbeil, J. Ray Meta: Scalable de novo metagenome assembly and profiling. Genome Biol. 2012, 13, R122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Y.; Leung, H.C.M.; Yiu, S.M.; Chin, F.Y.L. IDBA—A Practical Iterative de Bruijn Graph De Novo Assembler. In RECOMB 2010: Research in Computational Molecular Biology; Berger, B., Ed.; Lecture Notes in Computer Science; Springer: Berlin/Heidelberg, Germany, 2010; Volume 6044. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.; Leung, H.C.M.; Yiu, S.M.; Chin, F.Y.L. IDBA-UD: A de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 2012, 28, 1420–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. J. Comput. Mol. Cell Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nurk, S.; Meleshko, D.; Korobeynikov, A.; Pevzner, P.A. metaSPAdes: A new versatile metagenomic assembler. Genome Res. 2017, 27, 824–834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sczyrba, A.; Hofmann, P.; Belmann, P.; Koslicki, D.; Janssen, S.; Dröge, J.; Gregor, I.; Majda, S.; Fiedler, J.; Dahms, E.; et al. Critical Assessment of Metagenome Interpretation—A benchmark of metagenomics software. Nat. Methods 2017, 14, 1063–1071. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Liu, C.-M.; Luo, R.; Sadakane, K.; Lam, T.-W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagarajan, N.; Pop, M. Sequence assembly demystified. Nat. Rev. Genet. 2013, 14, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Koren, S.; Phillippy, A.M. One chromosome, one contig: Complete microbial genomes from long-read sequencing and assembly. Curr. Opin. Microbiol. 2015, 23, 110–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nobu, M.K.; Narihiro, T.; Rinke, C.; Kamagata, Y.; Tringe, S.; Woyke, T.; Liu, W.-T. Microbial dark matter ecogenomics reveals complex synergistic networks in a methanogenic bioreactor. ISME J. 2015, 9, 1710–1722. [Google Scholar] [CrossRef] [Green Version]
- Driscoll, C.B.; Otten, T.G.; Brown, N.M.; Dreher, T.W. Towards long-read metagenomics: Complete assembly of three novel genomes from bacteria dependent on a diazotrophic cyanobacterium in a freshwater lake co-culture. Stand. Genom. Sci. 2017, 12, 9. [Google Scholar] [CrossRef] [Green Version]
- Land, M.; Hauser, L.; Jun, S.-R.; Nookaew, I.; Leuze, M.R.; Ahn, T.-H.; Karpinets, T.; Lund, O.; Kora, G.; Wassenaar, T.; et al. Insights from 20 years of bacterial genome sequencing. Funct. Integr. Genom. 2015, 15, 141–161. [Google Scholar] [CrossRef] [Green Version]
- Schaeffer, L.; Pimentel, H.; Bray, N.; Melsted, P.; Pachter, L. Pseudoalignment for metagenomic read assignment. Bioinformatics 2017, 33, 2082–2088. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Benson, D.A.; Cavanaugh, M.; Clark, K.; Mizrachi, I.K.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2017, 45, D37–D42. [Google Scholar] [CrossRef] [Green Version]
- Baldrian, P.; López-Mondéjar, R. Microbial genomics, transcriptomics and proteomics: New discoveries in decomposition research using complementary methods. Appl. Microbiol. Biotechnol. 2014, 98, 1531–1537. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eddy, S.R. Accelerated Profile HMM Searches. PLoS Comput. Biol. 2011, 7, e1002195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darling, A.E.; Jospin, G.; Lowe, E.; Matsen, F.A., IV; Bik, H.M.; Eisen, J.A. PhyloSift: Phylogenetic analysis of genomes and metagenomes. PeerJ 2014, 2, e243. [Google Scholar] [CrossRef] [Green Version]
- Sunagawa, S.; Mende, D.R.; Zeller, G.; Izquierdo-Carrasco, F.; Berger, S.A.; Kultima, J.R.; Coelho, L.P.; Arumugam, M.; Tap, J.; Nielsen, H.B.; et al. Metagenomic species profiling using universal phylogenetic marker genes. Nat. Methods 2013, 10, 1196–1199. [Google Scholar] [CrossRef] [PubMed]
- Freitas, T.A.K.; Li, P.-E.; Scholz, M.; Chain, P.S.G. Accurate read-based metagenome characterization using a hierarchical suite of unique signatures. Nucleic Acids Res. 2015, 43, e69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [Green Version]
- Ondov, B.D.; Treangen, T.J.; Melsted, P.; Mallonee, A.B.; Bergman, N.H.; Koren, S.; Phillippy, A.M. Mash: Fast genome and metagenome distance estimation using MinHash. Genome Biol. 2016, 17, 132. [Google Scholar] [CrossRef] [Green Version]
- Brown, C.T.; Irber, L. sourmash: A library for MinHash sketching of DNA. J. Open Source Softw. 2016, 1, 27. [Google Scholar] [CrossRef] [Green Version]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Amir, A.; McDonald, D.; Navas-Molina, J.A.; Kopylova, E.; Morton, J.T.; Zech Xu, Z.; Kightley, E.P.; Thompson, L.R.; Hyde, E.R.; Gonzalez, A.; et al. Deblur Rapidly Resolves Single-Nucleotide Community Sequence Patterns. mSystems 2017, 2, e00191-16. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Kaehler, B.D.; Rideout, J.R.; Dillon, M.; Bolyen, E.; Knight, R.; Huttley, G.A.; Gregory Caporaso, J. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2′s q2-feature-classifier plugin. Microbiome 2018, 6, 90. [Google Scholar] [CrossRef] [PubMed]
- Janssen, S.; McDonald, D.; Gonzalez, A.; Navas-Molina, J.A.; Jiang, L.; Xu, Z.Z.; Winker, K.; Kado, D.M.; Orwoll, E.; Manary, M.; et al. Phylogenetic Placement of Exact Amplicon Sequences Improves Associations with Clinical Information. mSystems 2018, 3, e00021-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagalakshmi, U.; Wang, Z.; Waern, K.; Shou, C.; Raha, D.; Gerstein, M.; Snyder, M. The Transcriptional Landscape of the Yeast Genome Defined by RNA Sequencing. Science 2008, 320, 1344–1349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Cloonan, N.; Forrest, A.R.; Kolle, G.; Gardiner, B.B.; Faulkner, G.J.; Brown, M.K.; Taylor, D.F.; Steptoe, A.L.; Wani, S.; Bethel, G.; et al. Stem cell transcriptome profiling via massive-scale mRNA sequencing. Nat. Methods 2008, 5, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Cloonan, N.; Grimmond, S.M. Transcriptome content and dynamics at single-nucleotide resolution. Genome Biol. 2008, 9, 234. [Google Scholar] [CrossRef]
- Huson, D.H.; Richter, D.C.; Mitra, S.; Auch, A.F.; Schuster, S.C. Methods for comparative metagenomics. BMC Bioinform. 2009, 10, S12. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Brito, B.; Rohwer, F.; Edwards, R.A. An application of statistics to comparative metagenomics. BMC Bioinform. 2006, 7, 162. [Google Scholar] [CrossRef] [Green Version]
- Tringe, S.G.; von Mering, C.; Kobayashi, A.; Salamov, A.A.; Chen, K.; Chang, H.W.; Podar, M.; Short, J.M.; Mathur, E.J.; Detter, J.C.; et al. Comparative Metagenomics of Microbial Communities. Science 2005, 308, 554–557. [Google Scholar] [CrossRef] [Green Version]
- Nicolae, M.; Mangul, S.; Măndoiu, I.I.; Zelikovsky, A. Estimation of alternative splicing isoform frequencies from RNA-Seq data. Algorithms Mol. Biol. 2011, 6, 9. [Google Scholar] [CrossRef] [Green Version]
- Huson, D.H.; Auch, A.F.; Qi, J.; Schuster, S.C. MEGAN analysis of metagenomic data. Genome Res. 2007, 17, 377–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, C.; Cram, J.; Chen, T.; Fuhrman, J.; Sun, F. Accurate Genome Relative Abundance Estimation Based on Shotgun Metagenomic Reads. PLoS ONE 2011, 6, e27992. [Google Scholar] [CrossRef] [PubMed]
- Lindner, M.S.; Renard, B.Y. Metagenomic abundance estimation and diagnostic testing on species level. Nucleic Acids Res. 2013, 41, e10. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Knight, R.; Siljander, H.; Knip, M.; Xavier, R.J.; Gevers, D. ConStrains identifies microbial strains in metagenomic datasets. Nat. Biotechnol. 2015, 33, 1045–1052. [Google Scholar] [CrossRef] [Green Version]
- Sohn, M.B.; An, L.; Pookhao, N.; Li, Q. Accurate genome relative abundance estimation for closely related species in a metagenomic sample. BMC Bioinform. 2014, 15, 242. [Google Scholar] [CrossRef] [Green Version]
- Wood, D.E.; Salzberg, S.L. Kraken: Ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014, 15, R46. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Salzberg, S.L. Ultrafast and accurate 16S rRNA microbial community analysis using Kraken 2. Microbiome 2020, 8, 124. [Google Scholar] [CrossRef]
- Lu, J.; Breitwieser, F.P.; Thielen, P.; Salzberg, S.L. Bracken: Estimating species abundance in metagenomics data. PeerJ Comput. Sci. 2017, 3, e104. [Google Scholar] [CrossRef] [Green Version]
- Ma, B.; Tromp, J.; Li, M. PatternHunter: Faster and more sensitive homology search. Bioinformatics 2002, 18, 440–445. [Google Scholar] [CrossRef] [Green Version]
- Noé, L.; Martin, D.E. A Coverage Criterion for Spaced Seeds and Its Applications to Support Vector Machine String Kernels and k-Mer Distances. J. Comput. Biol. J. Comput. Mol. Cell Biol. 2014, 21, 947–963. [Google Scholar] [CrossRef]
- Kiełbasa, S.M.; Wan, R.; Sato, K.; Horton, P.; Frith, M.C. Adaptive seeds tame genomic sequence comparison. Genome Res. 2011, 21, 487–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef] [PubMed]
- Ainsworth, D.; Sternberg, M.J.; Raczy, C.; Butcher, S.A. k-SLAM: Accurate and ultra-fast taxonomic classification and gene identification for large metagenomic data sets. Nucleic Acids Res. 2017, 45, 1649–1656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2014, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Menzel, P.; Ng, K.L.; Krogh, A. Fast and sensitive taxonomic classification for metagenomics with Kaiju. Nat. Commun. 2016, 7, 11257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef] [Green Version]
- BBTools. Available online: https://jgi.doe.gov/data-and-tools/software-tools/bbtools/ (accessed on 19 October 2022).
- Crusoe, M.R.; Alameldin, H.F.; Awad, S.; Boucher, E.; Caldwell, A.; Cartwright, R.; Charbonneau, A.; Constantinides, B.; Edvenson, G.; Fay, S.; et al. The khmer software package: Enabling efficient nucleotide sequence analysis. F1000Research 2015, 4, 900. [Google Scholar] [CrossRef] [Green Version]
- Brown, C.T.; Howe, A.; Zhang, Q.; Pyrkosz, A.B.; Brom, T.H. A Reference-Free Algorithm for Computational Normalization of Shotgun Sequencing Data. arXiv 2012, arXiv:1203.4802. [Google Scholar]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [Green Version]
- Truong, D.; Franzosa, E.; Tickle, T.; Scholz, M.; Weingart, G.; Pasolli, E.; Tett, A.; Huttenhower, C.; Segata, N. MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat. Methods 2015, 12, 902–903. [Google Scholar] [CrossRef]
- Ounit, R.; Wanamaker, S.; Close, T.J.; Lonardi, S. CLARK: Fast and accurate classification of metagenomic and genomic sequences using discriminative k-mers. BMC Genom. 2015, 16, 236. [Google Scholar] [CrossRef] [PubMed]
- Hiltemann, S.; Batut, B.; Clements, D. 16S Microbial Analysis with Mothur (Extended). Available online: https://training.galaxyproject.org/training-material/topics/metagenomics/tutorials/mothur-miseq-sop/tutorial.html (accessed on 25 October 2022).
- Schloss, P.D. Reintroducing mothur: 10 Years Later. Appl. Environ. Microbiol. 2020, 86, e02343-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mothur Website. Available online: https://mothur.org (accessed on 25 October 2022).
- Westreich, S.T.; Treiber, M.L.; Mills, D.A.; Korf, I.; Lemay, D.G. SAMSA2: A standalone metatranscriptome analysis pipeline. BMC Bioinform. 2018, 19, 175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Tommaso, P.; Chatzou, M.; Floden, E.W.; Barja, P.P.; Palumbo, E.; Notredame, C. Nextflow enables reproducible computational workflows. Nat. Biotechnol. 2017, 35, 316–319. [Google Scholar] [CrossRef] [PubMed]
- Mende, D.R.; Waller, A.S.; Sunagawa, S.; Järvelin, A.I.; Chan, M.M.; Arumugam, M.; Raes, J.; Bork, P. Assessment of Metagenomic Assembly Using Simulated Next Generation Sequencing Data. PLoS ONE 2012, 7, e31386. Available online: https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0031386 (accessed on 20 October 2022). [CrossRef] [Green Version]
- Almeida, A.; Mitchell, A.L.; Tarkowska, A.; Finn, R.D. Benchmarking taxonomic assignments based on 16S rRNA gene profiling of the microbiota from commonly sampled environments. GigaScience 2018, 7, giy054. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Gu, Y.; He, Q.; Huang, J.; Song, Y.; Wan, X.; Li, Y. Integrated Analysis of Microbiome and Transcriptome Data Reveals the Interplay Between Commensal Bacteria and Fibrin Degradation in Endometrial Cancer. Front. Cell. Infect. Microbiol. 2021, 11, 748558. [Google Scholar] [CrossRef]
- Paulson, J.N.; Stine, O.C.; Bravo, H.C.; Pop, M. Differential abundance analysis for microbial marker-gene surveys. Nat. Methods 2013, 10, 1200–1202. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Lee, S.; Park, T. A Comparison Study of Statistical Methods for the Analysis Metagenome Data. In Proceedings of the 2017 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), Kansas City, MO, USA, 13 November 2017; pp. 1777–1781. [Google Scholar]
- Yoo, A.B.; Jette, M.A.; Grondona, M. SLURM: Simple Linux Utility for Resource Management. In Proceedings of the Job Scheduling Strategies for Parallel Processing, Santa Barbara, CA, USA, 25 April 2003; Feitelson, D., Rudolph, L., Schwiegelshohn, U., Eds.; Springer: Berlin/Heidelberg, Germany, 2003; pp. 44–60. [Google Scholar]
- AWS|Cloud Computing. Available online: https://aws.amazon.com/es/ (accessed on 26 October 2022).
- Kassambara, A. Ggpubr: “ggplot2” Based Publication Ready Plots 2020. Available online: https://github.com/kassambara/ggpubr (accessed on 28 November 2022).
- Lu, W.; He, F.; Lin, Z.; Liu, S.; Tang, L.; Huang, Y.; Hu, Z. Dysbiosis of the endometrial microbiota and its association with inflammatory cytokines in endometrial cancer. Int. J. Cancer 2021, 148, 1708–1716. [Google Scholar] [CrossRef]
- Ackerman, J. The Ultimate Social Network. Sci. Am. 2012, 306, 36–43. [Google Scholar] [CrossRef]
- Lindgreen, S.; Adair, K.L.; Gardner, P.P. An evaluation of the accuracy and speed of metagenome analysis tools. Sci. Rep. 2016, 6, 19233. [Google Scholar] [CrossRef] [PubMed]
- Ye, S.H.; Siddle, K.J.; Park, D.J.; Sabeti, P.C. Benchmarking Metagenomics Tools for Taxonomic Classification. Cell 2019, 178, 779–794. [Google Scholar] [CrossRef] [PubMed]
- Blauwkamp, T.A.; Thair, S.; Rosen, M.J.; Blair, L.; Lindner, M.S.; Vilfan, I.D.; Kawli, T.; Christians, F.C.; Venkatasubrahmanyam, S.; Wall, G.D.; et al. Analytical and clinical validation of a microbial cell-free DNA sequencing test for infectious disease. Nat. Microbiol. 2019, 4, 663–674. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Crawford, E.D.; O’Donovan, B.D.; Wilson, M.R.; Chow, E.D.; Retallack, H.; DeRisi, J.L. Depletion of Abundant Sequences by Hybridization (DASH): Using Cas9 to remove unwanted high-abundance species in sequencing libraries and molecular counting applications. Genome Biol. 2016, 17, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zinter, M.S.; Mayday, M.Y.; Ryckman, K.K.; Jelliffe-Pawlowski, L.L.; DeRisi, J.L. Towards precision quantification of contamination in metagenomic sequencing experiments. Microbiome 2019, 7, 62. [Google Scholar] [CrossRef]
- Rosselló-Móra, R.; Amann, R. Past and future species definitions for Bacteria and Archaea. Syst. Appl. Microbiol. 2015, 38, 209–216. [Google Scholar] [CrossRef]
- Lan, R.; Reeves, P.R. Escherichia coli in disguise: Molecular origins of Shigella. Microbes Infect. 2002, 4, 1125–1132. [Google Scholar] [CrossRef]
- Taylor, J.W. One Fungus = One Name: DNA and fungal nomenclature twenty years after PCR. IMA Fungus 2011, 2, 113–120. [Google Scholar] [CrossRef] [Green Version]
- Federhen, S. Type material in the NCBI Taxonomy Database. Nucleic Acids Res. 2015, 43, D1086–D1098. [Google Scholar] [CrossRef] [Green Version]
- Roux, S.; Hallam, S.J.; Woyke, T.; Sullivan, M.B. Viral dark matter and virus–host interactions resolved from publicly available microbial genomes. eLife 2015, 4, e08490. [Google Scholar] [CrossRef]
- Simmonds, P.; Adams, M.J.; Benkő, M.; Breitbart, M.; Brister, J.R.; Carstens, E.B.; Davison, A.J.; Delwart, E.; Gorbalenya, A.E.; Harrach, B.; et al. Consensus Statement: Virus Taxonomy in the Age of Metagenomics. Nat. Rev. Microbiol. 2017, 15, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P. Methods for virus classification and the challenge of incorporating metagenomic sequence data. J. Gen. Virol. 2015, 96, 1193–1206. [Google Scholar] [CrossRef] [PubMed]
- Tao, T.; Madden, T.; Christiam, C. BLAST FTP Site; National Center for Biotechnology Information (US): Verenigde Staten, MD, USA, 2020. [Google Scholar]
- Tatusova, T.; Ciufo, S.; Federhen, S.; Fedorov, B.; McVeigh, R.; O’Neill, K.; Tolstoy, I.; Zaslavsky, L. Update on RefSeq microbial genomes resources. Nucleic Acids Res. 2015, 43, D599–D605. [Google Scholar] [CrossRef] [PubMed]
- Richard, M.L.; Liguori, G.; Lamas, B.; Brandi, G.; DA Costa, G.; Hoffmann, T.W.; Di Simone, M.P.; Calabrese, C.; Poggioli, G.; Langella, P.; et al. Mucosa-associated microbiota dysbiosis in colitis associated cancer. Gut Microbes 2017, 9, 131–142. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Shao, L.; Liu, X.; Ji, F.; Mei, Y.; Cheng, Y.; Liu, F.; Yan, C.; Li, L.; Ling, Z. Alterations of gastric mucosal microbiota across different stomach microhabitats in a cohort of 276 patients with gastric cancer. eBioMedicine 2019, 40, 336–348. [Google Scholar] [CrossRef] [Green Version]
- Medina-Bastidas, D.; Camacho-Arroyo, I.; García-Gómez, E. Current findings in endometrial microbiome: Impact on uterine diseases. Reproduction 2022, 163, R81–R96. [Google Scholar] [CrossRef]













| Parameter | Function | Command |
|---|---|---|
| Confidence score threshold | kraken |
|
| Read length of the input data | bracken-build |
|
| kmer length of the reference database | bracken-build |
|
| Read length of the input data | bracken |
|
| Taxonomic level to filter by | bracken |
|
| Threshold for bracken filter | bracken |
|
| Parameter | Function | Command |
|---|---|---|
| Format of the input data (casava format, singled/paired, demultiplexed) | import |
|
| Position to trim reads | dada2 denoise |
|
| Position to truncate reads | dada2 denoise |
|
| Method used to remove chimeras | dada2 denoise |
|
| Frequency that each sample should be rarefied | diversity |
|
| Taxonomic level to filter by | collapse |
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Terrón-Camero, L.C.; Gordillo-González, F.; Salas-Espejo, E.; Andrés-León, E. Comparison of Metagenomics and Metatranscriptomics Tools: A Guide to Making the Right Choice. Genes 2022, 13, 2280. https://doi.org/10.3390/genes13122280
Terrón-Camero LC, Gordillo-González F, Salas-Espejo E, Andrés-León E. Comparison of Metagenomics and Metatranscriptomics Tools: A Guide to Making the Right Choice. Genes. 2022; 13(12):2280. https://doi.org/10.3390/genes13122280
Chicago/Turabian StyleTerrón-Camero, Laura C., Fernando Gordillo-González, Eduardo Salas-Espejo, and Eduardo Andrés-León. 2022. "Comparison of Metagenomics and Metatranscriptomics Tools: A Guide to Making the Right Choice" Genes 13, no. 12: 2280. https://doi.org/10.3390/genes13122280
APA StyleTerrón-Camero, L. C., Gordillo-González, F., Salas-Espejo, E., & Andrés-León, E. (2022). Comparison of Metagenomics and Metatranscriptomics Tools: A Guide to Making the Right Choice. Genes, 13(12), 2280. https://doi.org/10.3390/genes13122280