
A Patient with Corticobasal Syndrome and Progressive Non-Fluent Aphasia (CBS-PNFA), with Variants in ATP7B, SETX, SORL1, and FOXP1 Genes

 ,
,  , , , ,
, , , , 
Abstract
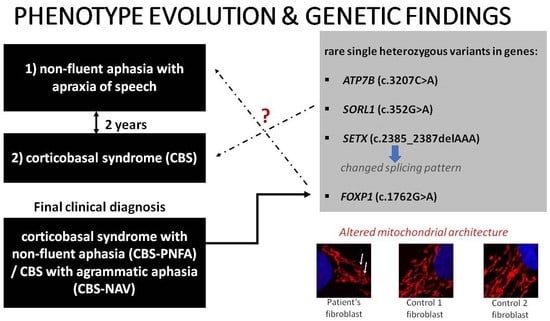
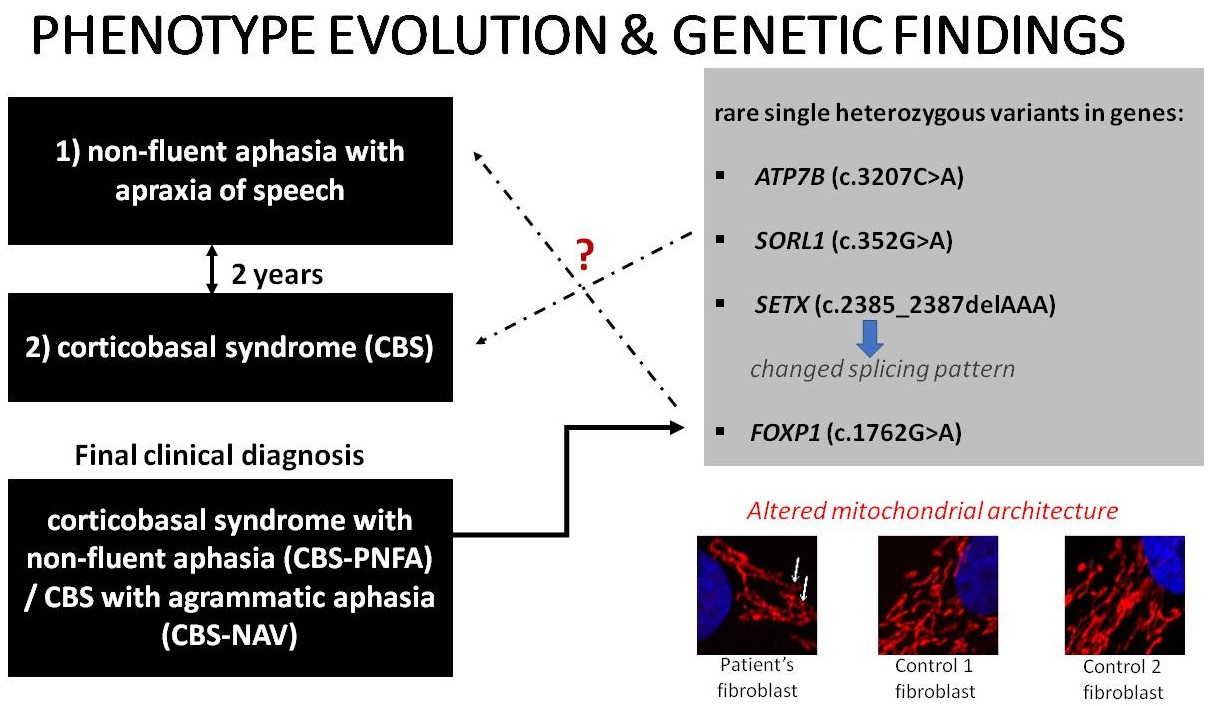
:
1. Introduction
2. Materials and Methods
2.1. Whole Exome Sequencing (WES) Data Preprocessing
2.2. Fibroblast Cultures and Inhibition of Nonsense-Mediated Decay (NMD)
2.3. Reverse Transcription, mRNA Isoform Analysis, and Real-Time PCR (RT-PCR)
2.4. Mitochondrial Network Staining and Analysis
2.5. Statistical Analysis
3. Results
3.1. Case Description
3.1.1. Clinical Assessment
3.1.2. Laboratory Testing
3.1.3. Neuroimaging
3.1.4. Genetic Assessment
3.1.5. Mitochondrial Network Assessment
4. Discussion
4.1. Complex Genetic Landscape
4.2. The significance of Motor Speech Disorders for Delineating the Patient’s Complex Clinical Phenotype
4.3. Altered Mitochondrial Architecture
4.4. Limitations of the Study
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ramos, E.M.; Dokuru, D.R.; Van Berlo, V.; Wojta, K.; Wang, Q.; Huang, A.Y.; Miller, Z.A.; Karydas, A.M.; Bigio, E.H.; Rogalski, E.; et al. Genetic screen in a large series of patients with primary progressive aphasia. Alzheimer’s Dement. 2019, 15, 553–560. [Google Scholar] [CrossRef] [PubMed]
- Arienti, F.; Lazzeri, G.; Vizziello, M.; Monfrini, E.; Bresolin, N.; Saetti, M.C.; Picillo, M.; Franco, G.; Di Fonzo, A. Unravelling Genetic Factors Underlying Corticobasal Syndrome: A Systematic Review. Cells 2021, 10, 171. [Google Scholar] [CrossRef] [PubMed]
- Bugiani, O.; Murrell, J.R.; Giaccone, G.; Hasegawa, M.; Ghigo, G.; Tabaton, M.; Morbin, M.; Primavera, A.; Carella, F.; Solaro, C.; et al. Frontotemporal dementia and corticobasal degeneration in a family with a P301S mutation in tau. J. Neuropathol. Exp. Neurol. 1999, 58, 667–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benussi, L.; Binetti, G.; Sina, E.; Gigola, L.; Bettecken, T.; Meitinger, T.; Ghidoni, R. A novel deletion in progranulin gene is associated with FTDP-17 and CBS. Neurobiol. Aging 2008, 29, 427–435. [Google Scholar] [CrossRef]
- Lindquist, S.G.; Duno, M.; Batbayli, M.; Puschmann, A.; Braendgaard, H.; Mardosiene, S.; Svenstrup, K.; Pinborg, L.H.; Vestergaard, K.; Hjermind, L.E.; et al. Corticobasal and ataxia syndromes widen the spectrum of C9ORF72 hexanucleotide expansion disease. Clin. Genet. 2013, 83, 279–283. [Google Scholar] [CrossRef] [PubMed]
- Chen-Plotkin, A.S.; Yuan, W.; Anderson, C.; McCarty Wood, E.; Hurtig, H.I.; Clark, C.M.; Miller, B.L.; Lee, V.M.; Trojanowski, J.Q.; Grossman, M.; et al. Corticobasal syndrome and primary progressive aphasia as manifestations of LRRK2 gene mutations. Neurology 2008, 70, 521–527. [Google Scholar] [CrossRef] [Green Version]
- Constantinides, V.C.; Paraskevas, G.P.; Paraskevas, P.G.; Stefanis, L.; Kapaki, E. Corticobasal degeneration and corticobasal syndrome: A review. Clin. Park. Relat. Disord. 2019, 1, 66–71. [Google Scholar] [CrossRef]
- Koga, S.; Josephs, K.A.; Aiba, I.; Yoshida, M.; Dickson, D.W. Neuropathology and emerging biomarkers in corticobasal syndrome. J. Neurol. Neurosurg. Psychiatry 2022, 93, 919–929. [Google Scholar] [CrossRef]
- Houlden, H.; Baker, M.; Morris, H.R.; MacDonald, N.; Pickering-Brown, S.; Adamson, J.; Lees, A.J.; Rossor, M.N.; Quinn, N.P.; Kertesz, A.; et al. Corticobasal degeneration and progressive supranuclear palsy share a common tau haplotype. Neurology 2001, 56, 1702–1706. [Google Scholar] [CrossRef]
- Kouri, N.; Ross, O.A.; Dombroski, B.; Younkin, C.S.; Serie, D.J.; Soto-Ortolaza, A.; Baker, M.; Finch, N.C.A.; Yoon, H.; Kim, J.; et al. Genome-wide association study of corticobasal degeneration identifies risk variants shared with progressive supranuclear palsy. Nat. Commun. 2015, 6, 7247. [Google Scholar] [CrossRef]
- Borroni, B.; Pilotto, A.; Bianchi, M.; Gilberti, N.; Padovani, A. Genetic contributors to frontotemporal lobar degeneration: Beyond monogenic disease. Mini Rev. Med. Chem. 2011, 11, 988–1001. [Google Scholar] [CrossRef] [PubMed]
- Snowden, J.S.; Adams, J.; Harris, J.; Thompson, J.C.; Rollinson, S.; Richardson, A.; Jones, M.; Neary, D.; Mann, D.M.; Pickering-Brown, S. Distinct clinical and pathological phenotypes in frontotemporal dementia associated with MAPT, PGRN and C9orf72 mutations. Amyotroph. Lateral Scler. Front. Degener. 2015, 16, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Gossye, H.; Van Broeckhoven, C.; Engelborghs, S. The Use of Biomarkers and Genetic Screening to Diagnose Frontotemporal Dementia: Evidence and Clinical Implications. Front. Neurosci. 2019, 13, 757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snowden, J.S.; Pickering-Brown, S.M.; Mackenzie, I.R.; Richardson, A.M.; Varma, A.; Neary, D.; Mann, D.M. Progranulin gene mutations associated with frontotemporal dementia and progressive non-fluent aphasia. Brain 2006, 129, 3091–3102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harciarek, M.; Sitek, E.J.; Kertesz, A. The patterns of progression in primary progressive aphasia—Implications for assessment and management. Aphasiology 2014, 28, 964–980. [Google Scholar] [CrossRef]
- Kertesz, A.; McMonagle, P.; Blair, M.; Davidson, W.; Munoz, D.G. The evolution and pathology of frontotemporal dementia. Brain 2005, 128, 1996–2005. [Google Scholar] [CrossRef] [Green Version]
- Josephs, K.A.; Duffy, J.R. Apraxia of speech and nonfluent aphasia: A new clinical marker for corticobasal degeneration and progressive supranuclear palsy. Curr. Opin. Neurol. 2008, 21, 688–692. [Google Scholar] [CrossRef]
- Armstrong, M.J.; Litvan, I.; Lang, A.E.; Bak, T.H.; Bhatia, K.P.; Borroni, B.; Boxer, A.L.; Dickson, D.W.; Grossman, M.; Hallett, M.; et al. Criteria for the diagnosis of corticobasal degeneration. Neurology 2013, 80, 496–503. [Google Scholar] [CrossRef] [Green Version]
- Wilson, D.; Le Heron, C.; Anderson, T. Corticobasal syndrome: A practical guide. Pract. Neurol. 2021, 21, 276–285. [Google Scholar] [CrossRef]
- Dopper, E.G.; Seelaar, H.; Chiu, W.Z.; de Koning, I.; van Minkelen, R.; Baker, M.C.; Rozemuller, A.J.; Rademakers, R.; van Swieten, J.C. Symmetrical corticobasal syndrome caused by a novel C.314dup progranulin mutation. J. Mol. Neurosci. 2011, 45, 354–358. [Google Scholar] [CrossRef]
- Marshall, C.R.; Guerreiro, R.; Thust, S.; Fletcher, P.; Rohrer, J.D.; Fox, N.C. A Novel MAPT Mutation Causing Corticobasal Syndrome Led by Progressive Apraxia of Speech. J. Alzheimer’s Dis. 2015, 48, 923–926. [Google Scholar] [CrossRef] [PubMed]
- Mazzon, G.; Menichelli, A.; Fabretto, A.; Cattaruzza, T.; Manganotti, P. A new MAPT deletion in a case of speech apraxia leading to corticobasal syndrome. Neurocase 2018, 24, 140–144. [Google Scholar] [CrossRef]
- Santos-Santos, M.A.; Mandelli, M.L.; Binney, R.J.; Ogar, J.; Wilson, S.M.; Henry, M.L.; Hubbard, H.I.; Meese, M.; Attygalle, S.; Rosenberg, L.; et al. Features of Patients With Nonfluent/Agrammatic Primary Progressive Aphasia With Underlying Progressive Supranuclear Palsy Pathology or Corticobasal Degeneration. JAMA Neurol. 2016, 73, 733–742. [Google Scholar] [CrossRef] [PubMed]
- Katsanis, N. The continuum of causality in human genetic disorders. Genome Biol. 2016, 17, 233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabuas-Pereira, M.; Guerreiro, R.; Kun-Rodrigues, C.; Almeida, M.R.; Bras, J.; Santana, I. Whole-exome sequencing reveals PSEN1 and ATP7B combined variants as a possible cause of early-onset Lewy body dementia: A case study of genotype-phenotype correlation. Neurogenetics 2022, 23, 279–283. [Google Scholar] [CrossRef]
- Gaweda-Walerych, K.; Sitek, E.J.; Borczyk, M.; Berdynski, M.; Narozanska, E.; Brockhuis, B.; Korostynski, M.; Slawek, J.; Zekanowski, C. Two Rare Variants in PLAU and BACE1 Genes-Do They Contribute to Semantic Dementia Clinical Phenotype? Genes 2021, 12, 1806. [Google Scholar] [CrossRef]
- Cochran, J.N.; McKinley, E.C.; Cochran, M.; Amaral, M.D.; Moyers, B.A.; Lasseigne, B.N.; Gray, D.E.; Lawlor, J.M.J.; Prokop, J.W.; Geier, E.G.; et al. Genome sequencing for early-onset or atypical dementia: High diagnostic yield and frequent observation of multiple contributory alleles. Mol. Case Stud. 2019, 5, a003491. [Google Scholar] [CrossRef] [Green Version]
- Ciani, M.; Bonvicini, C.; Scassellati, C.; Carrara, M.; Maj, C.; Fostinelli, S.; Binetti, G.; Ghidoni, R.; Benussi, L. The Missing Heritability of Sporadic Frontotemporal Dementia: New Insights from Rare Variants in Neurodegenerative Candidate Genes. Int. J. Mol. Sci. 2019, 20, 3903. [Google Scholar] [CrossRef] [Green Version]
- Svenningsson, P. Corticobasal degeneration: Advances in clinicopathology and biomarkers. Curr. Opin. Neurol. 2019, 32, 597–603. [Google Scholar] [CrossRef]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef]
- Gaweda-Walerych, K.; Sitek, E.J.; Narozanska, E.; Wezyk, M.; Brockhuis, B.; Zekanowski, C.; Slawek, J. Functional characterization of a novel progranulin mutation in a patient with progressive nonfluent aphasia. Neurobiol. Aging 2018, 72, 186-e9. [Google Scholar] [CrossRef] [PubMed]
- Leparc, G.G.; Mitra, R.D. A sensitive procedure to detect alternatively spliced mRNA in pooled-tissue samples. Nucleic Acids Res. 2007, 35, e146. [Google Scholar] [CrossRef] [PubMed]
- Wauters, F.; Cornelissen, T.; Imberechts, D.; Martin, S.; Koentjoro, B.; Sue, C.; Vangheluwe, P.; Vandenberghe, W. LRRK2 mutations impair depolarization-induced mitophagy through inhibition of mitochondrial accumulation of RAB10. Autophagy 2020, 16, 203–222. [Google Scholar] [CrossRef] [PubMed]
- Merrill, K.H.F.; Strack, S. Measuring Mitochondrial Shape with ImageJ. In Techniques to Investigate Mitochondrial Function in Neurons; Strack, S., Usachev, Y.M., Eds.; Springer: Berlin/Heidelberg, Germany, 2017. [Google Scholar]
- Durand, M.J.; Ait-Aissa, K.; Levchenko, V.; Staruschenko, A.; Gutterman, D.D.; Beyer, A.M. Visualization and quantification of mitochondrial structure in the endothelium of intact arteries. Cardiovasc. Res. 2019, 115, 1546–1556. [Google Scholar] [CrossRef]
- Onesto, E.; Colombrita, C.; Gumina, V.; Borghi, M.O.; Dusi, S.; Doretti, A.; Fagiolari, G.; Invernizzi, F.; Moggio, M.; Tiranti, V.; et al. Gene-specific mitochondria dysfunctions in human TARDBP and C9ORF72 fibroblasts. Acta Neuropathol. Commun. 2016, 4, 47. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef]
- Wittmann, J.; Hol, E.M.; Jack, H.M. hUPF2 silencing identifies physiologic substrates of mammalian nonsense-mediated mRNA decay. Mol. Cell Biol. 2006, 26, 1272–1287. [Google Scholar] [CrossRef] [Green Version]
- Gong, C.; Kim, Y.K.; Woeller, C.F.; Tang, Y.; Maquat, L.E. SMD and NMD are competitive pathways that contribute to myogenesis: Effects on PAX3 and myogenin mRNAs. Genes Dev. 2009, 23, 54–66. [Google Scholar] [CrossRef] [Green Version]
- Barthelson, K.; Pederson, S.M.; Newman, M.; Lardelli, M. Brain transcriptome analysis reveals subtle effects on mitochondrial function and iron homeostasis of mutations in the SORL1 gene implicated in early onset familial Alzheimer’s disease. Mol. Brain 2020, 13, 142. [Google Scholar] [CrossRef]
- Renaudin, X.; Venkitaraman, A.R. A mitochondrial response to oxidative stress mediated by unscheduled RNA-DNA hybrids (R-loops). Mol. Cell Oncol. 2021, 8, 2007028. [Google Scholar] [CrossRef] [PubMed]
- Dev, S.; Kruse, R.L.; Hamilton, J.P.; Lutsenko, S. Wilson Disease: Update on Pathophysiology and Treatment. Front. Cell Dev. Biol. 2022, 10, 871877. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Frohlich, H.; Torres, F.B.; Silva, R.L.; Poschet, G.; Agarwal, A.; Rappold, G.A. Mitochondrial dysfunction and oxidative stress contribute to cognitive and motor impairment in FOXP1 syndrome. Proc. Natl. Acad. Sci. USA 2022, 119, e2112852119. [Google Scholar] [CrossRef] [PubMed]
- Roberts, E.A.; Schilsky, M.L. A practice guideline on Wilson disease. Hepatology 2003, 37, 1475–1492. [Google Scholar] [CrossRef] [Green Version]
- Lutsenko, S. Modifying factors and phenotypic diversity in Wilson’s disease. Ann. N. Y. Acad. Sci. 2014, 1315, 56–63. [Google Scholar] [CrossRef] [Green Version]
- Bandmann, O.; Weiss, K.H.; Kaler, S.G. Wilson’s disease and other neurological copper disorders. Lancet Neurol. 2015, 14, 103–113. [Google Scholar] [CrossRef] [Green Version]
- Pfeiffer, R.F. Wilson Disease. Contin. Lifelong Learn. Neurol. 2016, 22, 1246–1261. [Google Scholar] [CrossRef]
- Shah, A.B.; Chernov, I.; Zhang, H.T.; Ross, B.M.; Das, K.; Lutsenko, S.; Parano, E.; Pavone, L.; Evgrafov, O.; Ivanova-Smolenskaya, I.A.; et al. Identification and analysis of mutations in the Wilson disease gene (ATP7B): Population frequencies, genotype-phenotype correlation, and functional analyses. Am. J. Hum. Genet. 1997, 61, 317–328. [Google Scholar] [CrossRef] [Green Version]
- Ivanova-Smolenskaya, I.A.; Ovchinnikov, I.V.; Karabanov, A.V.; Deineko, N.L.; Poleshchuk, V.V.; Markova, E.D.; Illarioshkin, S.N. The His1069Gln mutation in the ATP7B gene in Russian patients with Wilson disease. J. Med. Genet. 1999, 36, 174. [Google Scholar]
- Rodriguez-Granillo, A.; Sedlak, E.; Wittung-Stafshede, P. Stability and ATP binding of the nucleotide-binding domain of the Wilson disease protein: Effect of the common H1069Q mutation. J. Mol. Biol. 2008, 383, 1097–1111. [Google Scholar] [CrossRef]
- Gromadzka, G.; Schmidt, H.H.; Genschel, J.; Bochow, B.; Rodo, M.; Tarnacka, B.; Litwin, T.; Chabik, G.; Czlonkowska, A. Frameshift and nonsense mutations in the gene for ATPase7B are associated with severe impairment of copper metabolism and with an early clinical manifestation of Wilson’s disease. Clin. Genet. 2005, 68, 524–532. [Google Scholar] [CrossRef] [PubMed]
- Kucinskas, L.; Jeroch, J.; Vitkauskiene, A.; Sakalauskas, R.; Petrenkiene, V.; Kucinskas, V.; Naginiene, R.; Schmidt, H.; Kupcinskas, L. High frequency of the c.3207C>A (p.H1069Q) mutation in ATP7B gene of Lithuanian patients with hepatic presentation of Wilson’s disease. World J. Gastroenterol. 2008, 14, 5876–5879. [Google Scholar] [CrossRef] [PubMed]
- Tarnacka, B.; Szeszkowski, W.; Buettner, J.; Golebiowski, M.; Gromadzka, G.; Czlonkowska, A. Heterozygous carriers for Wilson’s disease--magnetic spectroscopy changes in the brain. Metab. Brain Dis. 2009, 24, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Skowronska, M.; Litwin, T.; Kurkowska-Jastrzebska, I.; Czlonkowska, A. Transcranial sonography changes in heterozygotic carriers of the ATP7B gene. Neurol. Sci. 2020, 41, 2605–2612. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Lindauer, C.; Haase, R.; Rudiger, H.; Reichmann, H.; Reuner, U.; Ziemssen, T. Autonomic Dysfunction in Wilson’s Disease: A Comprehensive Evaluation during a 3-Year Follow Up. Front. Physiol. 2017, 8, 778. [Google Scholar] [CrossRef] [PubMed]
- Groh, M.; Albulescu, L.O.; Cristini, A.; Gromak, N. Senataxin: Genome Guardian at the Interface of Transcription and Neurodegeneration. J. Mol. Biol. 2017, 429, 3181–3195. [Google Scholar] [CrossRef] [PubMed]
- Richard, P.; Feng, S.; Tsai, Y.L.; Li, W.; Rinchetti, P.; Muhith, U.; Irizarry-Cole, J.; Stolz, K.; Sanz, L.A.; Hartono, S.; et al. SETX (senataxin), the helicase mutated in AOA2 and ALS4, functions in autophagy regulation. Autophagy 2021, 17, 1889–1906. [Google Scholar] [CrossRef]
- Grunseich, C.; Patankar, A.; Amaya, J.; Watts, J.A.; Li, D.; Ramirez, P.; Schindler, A.B.; Fischbeck, K.H.; Cheung, V.G. Clinical and Molecular Aspects of Senataxin Mutations in Amyotrophic Lateral Sclerosis 4. Ann. Neurol. 2020, 87, 547–555. [Google Scholar] [CrossRef] [Green Version]
- Hoyer, H.; Braathen, G.J.; Busk, O.L.; Holla, O.L.; Svendsen, M.; Hilmarsen, H.T.; Strand, L.; Skjelbred, C.F.; Russell, M.B. Genetic diagnosis of Charcot-Marie-Tooth disease in a population by next-generation sequencing. Biomed Res. Int. 2014, 2014, 210401. [Google Scholar] [CrossRef] [Green Version]
- Dierick, I.; Baets, J.; Irobi, J.; Jacobs, A.; De Vriendt, E.; Deconinck, T.; Merlini, L.; Van den Bergh, P.; Rasic, V.M.; Robberecht, W.; et al. Relative contribution of mutations in genes for autosomal dominant distal hereditary motor neuropathies: A genotype-phenotype correlation study. Brain 2008, 131, 1217–1227. [Google Scholar] [CrossRef] [Green Version]
- Worthey, E.A.; Raca, G.; Laffin, J.J.; Wilk, B.M.; Harris, J.M.; Jakielski, K.J.; Dimmock, D.P.; Strand, E.A.; Shriberg, L.D. Whole-exome sequencing supports genetic heterogeneity in childhood apraxia of speech. J. Neurodev. Disord. 2013, 5, 29. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Zhang, J.; Su, N.; Zhang, S.; Yan, F.; Lin, X.; Yu, J.; Li, W.; Li, X.; Xiao, S. Analysis of Genotype-Phenotype Correlations in Patients With Degenerative Dementia Through the Whole Exome Sequencing. Front. Aging Neurosci. 2021, 13, 745407. [Google Scholar] [CrossRef] [PubMed]
- Pros, E.; Larriba, S.; Lopez, E.; Ravella, A.; Gili, M.L.; Kruyer, H.; Valls, J.; Serra, E.; Lazaro, C. NF1 mutation rather than individual genetic variability is the main determinant of the NF1-transcriptional profile of mutations affecting splicing. Hum. Mutat. 2006, 27, 1104–1114. [Google Scholar] [CrossRef] [PubMed]
- Baralle, D.; Buratti, E. RNA splicing in human disease and in the clinic. Clin. Sci. 2017, 131, 355–368. [Google Scholar] [CrossRef]
- Pros, E.; Gomez, C.; Martin, T.; Fabregas, P.; Serra, E.; Lazaro, C. Nature and mRNA effect of 282 different NF1 point mutations: Focus on splicing alterations. Hum. Mutat. 2008, 29, E173–E193. [Google Scholar] [CrossRef]
- Raponi, M.; Smith, L.D.; Silipo, M.; Stuani, C.; Buratti, E.; Baralle, D. BRCA1 exon 11 a model of long exon splicing regulation. RNA Biol. 2014, 11, 351–359. [Google Scholar] [CrossRef] [Green Version]
- Willnow, T.E.; Andersen, O.M. Sorting receptor SORLA--a trafficking path to avoid Alzheimer disease. J. Cell Sci. 2013, 126, 2751–2760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, V.; Subkhangulova, A.; Willnow, T.E. Sorting receptor SORLA: Cellular mechanisms and implications for disease. Cell Mol. Life Sci. 2017, 74, 1475–1483. [Google Scholar] [CrossRef] [Green Version]
- Rogaeva, E.; Meng, Y.; Lee, J.H.; Gu, Y.; Kawarai, T.; Zou, F.; Katayama, T.; Baldwin, C.T.; Cheng, R.; Hasegawa, H.; et al. The neuronal sortilin-related receptor SORL1 is genetically associated with Alzheimer disease. Nat. Genet. 2007, 39, 168–177. [Google Scholar] [CrossRef]
- Bellenguez, C.; Charbonnier, C.; Grenier-Boley, B.; Quenez, O.; Le Guennec, K.; Nicolas, G.; Chauhan, G.; Wallon, D.; Rousseau, S.; Richard, A.C.; et al. Contribution to Alzheimer’s disease risk of rare variants in TREM2, SORL1, and ABCA7 in 1779 cases and 1273 controls. Neurobiol. Aging 2017, 59, 220.e1–220.e9. [Google Scholar] [CrossRef]
- Campion, D.; Charbonnier, C.; Nicolas, G. SORL1 genetic variants and Alzheimer disease risk: A literature review and meta-analysis of sequencing data. Acta Neuropathol. 2019, 138, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Campoy, O.; Antonell, A.; Falgas, N.; Balasa, M.; Borrego-Ecija, S.; Rodriguez-Santiago, B.; Datta, D.; Armengol, L.; Fernandez-Villullas, G.; Bosch, B.; et al. Screening of dementia genes by whole-exome sequencing in Spanish patients with early-onset dementia: Likely pathogenic, uncertain significance and risk variants. Neurobiol. Aging 2020, 93, e1–e9. [Google Scholar] [CrossRef] [PubMed]
- Sassi, C.; Capozzo, R.; Hammer, M.; Zecca, C.; Federoff, M.; Blauwendraat, C.; Bernstein, N.; Ding, J.; Gibbs, J.R.; Price, T.; et al. Exploring dementia and neuronal ceroid lipofuscinosis genes in 100 FTD-like patients from 6 towns and rural villages on the Adriatic Sea cost of Apulia. Sci. Rep. 2021, 11, 6353. [Google Scholar] [CrossRef] [PubMed]
- Csaban, D.; Illes, A.; Renata, T.B.; Balicza, P.; Pentelenyi, K.; Molnar, V.; Gezsi, A.; Grosz, Z.; Gal, A.; Kovacs, T.; et al. Genetic landscape of early-onset dementia in Hungary. Neurol. Sci. 2022, 43, 5289–5300. [Google Scholar] [CrossRef]
- Rovelet-Lecrux, A.; Feuillette, S.; Miguel, L.; Schramm, C.; Pernet, S.; Quenez, O.; Segalas-Milazzo, I.; Guilhaudis, L.; Rousseau, S.; Riou, G.; et al. Impaired SorLA maturation and trafficking as a new mechanism for SORL1 missense variants in Alzheimer disease. Acta Neuropathol. Commun. 2021, 9, 196. [Google Scholar] [CrossRef]
- Co, M.; Anderson, A.G.; Konopka, G. FOXP transcription factors in vertebrate brain development, function, and disorders. Wiley Interdiscip. Rev. Dev. Biol. 2020, 9, e375. [Google Scholar] [CrossRef]
- Braden, R.O.; Amor, D.J.; Fisher, S.E.; Mei, C.; Myers, C.T.; Mefford, H.; Gill, D.; Srivastava, S.; Swanson, L.C.; Goel, H.; et al. Severe speech impairment is a distinguishing feature of FOXP1-related disorder. Dev. Med. Child Neurol. 2021, 63, 1417–1426. [Google Scholar] [CrossRef]
- Siper, P.M.; De Rubeis, S.; Trelles, M.D.P.; Durkin, A.; Di Marino, D.; Muratet, F.; Frank, Y.; Lozano, R.; Eichler, E.E.; Kelly, M.; et al. Prospective investigation of FOXP1 syndrome. Mol. Autism 2017, 8, 57. [Google Scholar] [CrossRef] [Green Version]
- Bacon, C.; Rappold, G.A. The distinct and overlapping phenotypic spectra of FOXP1 and FOXP2 in cognitive disorders. Hum. Genet. 2012, 131, 1687–1698. [Google Scholar] [CrossRef] [Green Version]
- Padovani, A.; Cosseddu, M.; Premi, E.; Archetti, S.; Papetti, A.; Agosti, C.; Bigni, B.; Cerini, C.; Paghera, B.; Bellelli, G.; et al. The speech and language FOXP2 gene modulates the phenotype of frontotemporal lobar degeneration. J. Alzheimer’s Dis. 2010, 22, 923–931. [Google Scholar] [CrossRef]
- Molloy, J.; Jagoe, C. Use of diverse diagnostic criteria for acquired apraxia of speech: A scoping review. Int. J. Lang. Commun. Disord. 2019, 54, 875–893. [Google Scholar] [CrossRef] [PubMed]
- Jauer-Niworowska. A specific picture of speech disturbances in polish speaking patients with mixed dysarthria in multiple sclerosis (MS) and in Wilson’s disease (WD). Acta Neuropsychol. 2014, 12, 155–166. [Google Scholar]
- Poujois, A.; Pernon, M.; Trocello, J.M.; Woimant, F. Dystonic Dysarthria in Wilson Disease: Efficacy of Zolpidem. Front. Neurol. 2017, 8, 559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ilyechova, E.Y.; Miliukhina, I.V.; Karpenko, M.N.; Orlov, I.A.; Puchkova, L.V.; Samsonov, S.A. Case of Early-Onset Parkinson’s Disease in a Heterozygous Mutation Carrier of the ATP7B Gene. J. Pers. Med. 2019, 9, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruggeri, M.; Biagioli, C.; Ricci, M.; Gerace, C.; Blundo, C. Progressive aphasia, apraxia of speech and agraphia in corticobasal degeneration: A 12-case series clinical and neuropsychological descriptive study. Int. J. Lang. Commun. Disord. 2020, 55, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Tetzloff, K.A.; Duffy, J.R.; Strand, E.A.; Machulda, M.M.; Boland, S.M.; Utianski, R.L.; Botha, H.; Senjem, M.L.; Schwarz, C.G.; Josephs, K.A.; et al. Clinical and imaging progression over 10 years in a patient with primary progressive apraxia of speech and autopsy-confirmed corticobasal degeneration. Neurocase 2018, 24, 111–120. [Google Scholar] [CrossRef]
- Constantinides, V.C.; Souvatzoglou, M.; Paraskevas, G.P.; Chalioti, M.; Boufidou, F.; Stefanis, L.; Kapaki, E. Dopamine transporter SPECT imaging in corticobasal syndrome: A peak into the underlying pathology? Acta Neurol. Scand. 2022, 145, 762–769. [Google Scholar] [CrossRef]
- Nakano, Y.; Shimada, H.; Shinotoh, H.; Hirano, S.; Tagai, K.; Sano, Y.; Yamamoto, Y.; Endo, H.; Matsuoka, K.; Takahata, K.; et al. PET-based classification of corticobasal syndrome. Park. Relat. Disord. 2022, 98, 92–98. [Google Scholar] [CrossRef]
- Chung, S.W.; Choi, B.M.; Kim, J.Y.; Lee, Y.S.; Yoon, J.P.; Oh, K.S.; Park, K.S. Altered Gene and Protein Expressions in Torn Rotator Cuff Tendon Tissues in Diabetic Patients. Arthroscopy 2017, 33, 518–526.e1. [Google Scholar] [CrossRef]
- Lefever, S.; Vandesompele, J.; Speleman, F.; Pattyn, F. RTPrimerDB: The portal for real-time PCR primers and probes. Nucleic Acids Res. 2009, 37, D942–D945. [Google Scholar] [CrossRef] [Green Version]
- Manoochehri, J.; Masoumi Dehshiri, R.; Faraji, H.; Mohammadi, S.; Dastsooz, H.; Moradi, T.; Rezaei, E.; Sadeghi, K.; Fardaei, M. Family screening for a novel ATP7B gene mutation, c.2335T>G, in the South of Iran. Iran. J. Ped. Hematol. Oncol. 2014, 4, 26–31. [Google Scholar] [PubMed]
- Sechi, G.; Antonio Cocco, G.; Errigo, A.; Deiana, L.; Rosati, G.; Agnetti, V.; Stephen Paulus, K.; Mario Pes, G. Three sisters with very-late-onset major depression and parkinsonism. Park. Relat. Disord. 2007, 13, 122–125. [Google Scholar] [CrossRef] [PubMed]
- Moller, J.C.; Leinweber, B.; Rissling, I.; Oertel, W.H.; Bandmann, O.; Schmidt, H.H. Prevalence of the H1069Q mutation in ATP7B in discordant pairs with early-onset Parkinson’s disease. Mov. Disord. 2006, 21, 1789–1790. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Liu, J.; Zheng, W.; Hou, Q.; Zhang, L. A novel heterozygous carrier of ATP7B mutation with muscle weakness and tremor: A Chinese Case Report. J. Musculoskelet. Neuronal Interact. 2020, 20, 614–618. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number of Mapped Reads (in Millions) | Proportion of Sequenced Reads (in %) | PCR Duplicates (in %) * | Median Insert Size (in bp) | Average Coverage |
|---|---|---|---|---|
| 198.435 | 86.7 | 12.44 | 206 | 213.5 |
| Gene | HGVS 1 DNA/Exon/Protein/rs | Predicted Effect | MAF gnomAD | CADD * Phred | ACMG Criteria |
|---|---|---|---|---|---|
| ATP7B | c.3207C>A exon 14 p.His1069Gln rs76151636 heterozygous | missense | 0.001019 | 24 | PS1, BP4, BP5, PP3 likely pathogenic |
| SETX | c.2385_2387delAAA exon 10 p.Ile795_Lys796delinsMet rs755971927 heterozygous | in frame deletion | 0.0000329 | NA | PM2, PM4, PP4 uncertain significance |
| SORL1 | c.352G>A p.Val118Met exon 2 rs749389644 heterozygous | missense | 0.00001060 | 28.1 | PM2, PP3 uncertain significance |
| FOXP1 | c.1762G>A p.Ala588Thr exon 20 rs202173892 heterozygous | missense | 0.000318 | 21.7 | BP5 pathogenic/likely pathogenic or of uncertain impact |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gaweda-Walerych, K.; Sitek, E.J.; Borczyk, M.; Narożańska, E.; Brockhuis, B.; Korostyński, M.; Schinwelski, M.; Siemiński, M.; Sławek, J.; Zekanowski, C. A Patient with Corticobasal Syndrome and Progressive Non-Fluent Aphasia (CBS-PNFA), with Variants in ATP7B, SETX, SORL1, and FOXP1 Genes. Genes 2022, 13, 2361. https://doi.org/10.3390/genes13122361
Gaweda-Walerych K, Sitek EJ, Borczyk M, Narożańska E, Brockhuis B, Korostyński M, Schinwelski M, Siemiński M, Sławek J, Zekanowski C. A Patient with Corticobasal Syndrome and Progressive Non-Fluent Aphasia (CBS-PNFA), with Variants in ATP7B, SETX, SORL1, and FOXP1 Genes. Genes. 2022; 13(12):2361. https://doi.org/10.3390/genes13122361
Chicago/Turabian StyleGaweda-Walerych, Katarzyna, Emilia J. Sitek, Małgorzata Borczyk, Ewa Narożańska, Bogna Brockhuis, Michał Korostyński, Michał Schinwelski, Mariusz Siemiński, Jarosław Sławek, and Cezary Zekanowski. 2022. "A Patient with Corticobasal Syndrome and Progressive Non-Fluent Aphasia (CBS-PNFA), with Variants in ATP7B, SETX, SORL1, and FOXP1 Genes" Genes 13, no. 12: 2361. https://doi.org/10.3390/genes13122361
APA StyleGaweda-Walerych, K., Sitek, E. J., Borczyk, M., Narożańska, E., Brockhuis, B., Korostyński, M., Schinwelski, M., Siemiński, M., Sławek, J., & Zekanowski, C. (2022). A Patient with Corticobasal Syndrome and Progressive Non-Fluent Aphasia (CBS-PNFA), with Variants in ATP7B, SETX, SORL1, and FOXP1 Genes. Genes, 13(12), 2361. https://doi.org/10.3390/genes13122361